샷건 시퀀싱은 DNA 염기 서열 분석 방법으로, DNA를 무작위로 잘게 조각낸 후 각 조각의 염기 서열을 분석하여 전체 DNA 서열을 재구성한다. 1979년 처음 제안되었으며, 1981년 컬리플라워 모자이크 바이러스 게놈 분석에 처음 사용되었다. 페어 엔드 시퀀싱 기술의 발전으로 정확성과 효율성이 향상되었으며, 전체 유전체 샷건 시퀀싱과 계층적 샷건 시퀀싱 방법이 존재한다. 이 기술은 전체 유전체를 빠르게 시퀀싱할 수 있지만, 반복 서열이 많은 유전체는 분석에 어려움이 있다. 차세대 염기서열 분석 기술의 발전으로 더욱 빠르고 정확해졌으며, 메타게놈 샷건 시퀀싱을 통해 환경 미생물 군집 분석에도 활용된다. 한국에서는 질병 진단, 신약 개발 등 다양한 분야에서 활용되고 있다.
더 읽어볼만한 페이지
DNA 시퀀싱 - 차세대 염기서열 분석 차세대 염기서열 분석(NGS)은 대량의 DNA를 병렬적으로 분석하는 기술로, DNA 염기서열 라이브러리 준비, DNA 합성 기반 염기서열 결정, 대규모 병렬 염기서열 분석 등의 단계를 거쳐 수행되며, 생명 의학 분야의 유전체 염기서열 분석 방식을 혁신하고 분석 비용을 감소시켰다.
DNA 시퀀싱 - 총유전체 총유전체는 생물의 완전한 DNA 서열을 의미하며, 유전체 염기서열 분석을 통해 해독 가능하고, 자동화된 분석 방법 발전으로 분석이 용이해졌으며, 질병 예측, 맞춤형 치료 등 다양한 분야에 응용되지만 윤리적 문제와 개인 정보 보호가 중요하다.
샷건 시퀀싱
개요
유형
DNA 시퀀싱 방법
특징
DNA를 작은 조각으로 무작위 분할 후 시퀀싱 및 재조합
활용
전체 게놈 시퀀싱, 특정 DNA 영역 시퀀싱
상세 정보
원리
DNA를 작은 조각으로 쪼개어 개별적으로 염기서열을 분석한 다음, 컴퓨터 알고리즘을 사용하여 이 조각들을 원래 순서대로 재조합하여 전체 염기서열을 결정함.
과정
DNA 분할: DNA를 작은 조각으로 무작위로 분할 시퀀싱: 각 조각의 염기서열 결정 조립: 컴퓨터를 사용하여 조각들을 원래 순서대로 재조합
장점
시간 효율성: 기존 시퀀싱 방법에 비해 빠름 비용 효율성: 대량의 DNA 시퀀싱에 적합
단점
높은 계산 비용: 조립 과정에 많은 계산 자원 필요 반복 서열 문제: 반복적인 DNA 서열에서 오류 발생 가능성
역사
개발
프레더릭 생어가 개발한 체인 종결법 기반으로 발전
초기 적용
1980년대 초, 박테리오파지 λ의 게놈 시퀀싱에 사용
발전
컴퓨터 기술 발전으로 대규모 게놈 프로젝트에 적용 가능 인간 게놈 프로젝트에 중요한 역할
응용 분야
게놈 연구
인간 게놈 프로젝트 다양한 생물체의 게놈 해독
의학
질병 유전자 발견 개인 맞춤 의료
생물학
종의 진화 연구 생물 다양성 연구
법의학
DNA 감식을 통한 개인 식별
기술적 고려 사항
라이브러리 준비
시퀀싱할 DNA 단편들을 포함하는 라이브러리 제작
시퀀싱 깊이
각 DNA 단편을 여러 번 시퀀싱하여 정확도를 높임
데이터 분석
시퀀싱 결과 데이터를 분석하고 조립하는 과정
추가 정보
다른 이름
샷건 시퀀싱 방법
2. 역사
샷건 시퀀싱은 1979년 작은 게놈(4,000~7,000 염기쌍)에 대해 처음 제안되었다.[1] 1981년 컬리플라워 모자이크 바이러스의 게놈이 샷건 시퀀싱으로 해독되어 처음으로 발표되었다.[4][5]
2. 1. 페어 엔드 시퀀싱
페어 엔드 시퀀싱은 "더블 배럴 샷건 시퀀싱"이라고도 불리며, DNA 단편의 양쪽 끝을 염기서열 분석하여 정보를 얻는 방식이다. 이 방식은 염기서열 분석 프로젝트가 더 길고 복잡한 DNA 염기서열을 다루게 되면서 발전했다. 동일한 단편의 양쪽 끝 염기서열과 페어 데이터를 추적하는 것은 번거롭지만, 두 염기서열이 반대 방향으로 정렬되고 단편 길이만큼 떨어져 있다는 사실은 원래 목표 단편의 염기서열을 재구성하는 데 유용했다.
'''역사'''
페어 엔드 사용에 대한 최초의 출판된 설명은 1990년 인간 HGPRT 유전자좌의 염기서열 분석의 일부로 등장했다.[6] 하지만, 이때는 전통적인 샷건 염기서열 분석 후 갭을 닫는 데에만 제한적으로 사용되었다. 1991년에는 고정된 길이의 단편을 가정하는 순수한 페어 엔드 시퀀싱 전략에 대한 최초의 이론적 설명이 제시되었다.[7] 당시에는 페어 엔드 시퀀싱에 최적인 단편 길이는 염기서열 리드 길이의 3배여야 한다는 사회적 합의가 있었다. 1995년 로치 등은[8] 다양한 크기의 단편을 사용하는 혁신적인 방법을 도입하여, 순수한 페어 엔드 시퀀싱 전략이 대규모 목표에 적용될 수 있음을 보여주었다. 이 전략은 이후 1995년 TIGR에 의해 세균 ''Haemophilus influenzae''의 유전체 염기서열 분석에 채택되었고,[9] 2000년에는 Celera Genomics에 의해 ''Drosophila melanogaster''(초파리) 유전체, 이후 인간 유전체 염기서열 분석에 사용되었다.[10]
위의 예시에서 리드들은 원래 염기서열의 전체 길이를 포함하고 있지는 않지만, 네 개의 리드는 끝부분의 중첩을 활용하여 정렬 및 순서를 정함으로써 원래의 염기서열로 조합될 수 있다. 그러나 실제 상황에서는 이 과정은 수많은 불확실성과 시퀀싱 오류로 가득 차 있다. 복잡한 게놈의 조합은 반복 서열이 매우 풍부하기 때문에 더욱 복잡해진다. 이는 유사한 짧은 리드가 완전히 다른 부분에서 나올 수 있음을 의미한다.
이러한 어려움을 극복하고 정확한 시퀀스 조립을 위해서는 원래 DNA의 각 부분에 대해 많은 중첩 리드가 필요하다. 예를 들어, 인간 게놈 프로젝트를 완료하기 위해 인간 게놈의 대부분은 12X 또는 그 이상의 '커버리지'로 시퀀싱되었다. 즉, 최종 시퀀스의 각 염기는 평균적으로 12개의 다른 리드에 존재했다. 그럼에도 불구하고 현재의 방법으로는 2004년 기준으로 약 1%의 유크로마틴(사람 게놈)에 대한 신뢰할 수 있는 시퀀스를 분리하거나 조립하는 데 실패했다.[3]
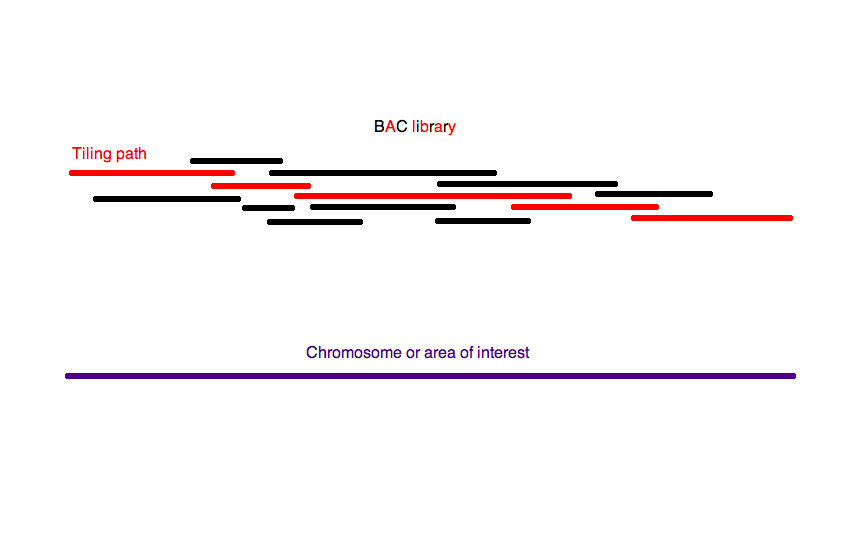
계층적 샷건 시퀀싱(top-down 시퀀싱이라고도 함)에서는 실제 시퀀싱 전에 유전체의 낮은 해상도 물리적 지도가 만들어진다.[15] 이 지도에서 전체 염색체를 덮는 최소 수의 단편이 시퀀싱을 위해 선택된다.[15] 이러한 방식으로 최소한의 고처리량 시퀀싱과 조립이 필요하다.
증폭된 유전체는 먼저 더 큰 조각(50-200kb)으로 분해되어 BAC 또는 P1 유래 인공 염색체(PAC)를 사용하여 세균 숙주에 클론된다. 여러 유전체 사본이 무작위로 분해되었기 때문에 이러한 클론에 포함된 단편은 서로 다른 끝을 가지며, 충분한 커버리지를 통해 전체 유전체를 덮는 가장 작은 '''스캐폴드''' BAC 콘티그를 찾는 것이 이론적으로 가능하다. 이 스캐폴드를 '''최소 타일링 경로'''라고 한다. 관심 영역의 전체 유전체를 덮는 BAC 콘티그가 타일링 경로를 구성한다. 타일링 경로를 찾으면 이 경로를 형성하는 BAC가 무작위로 더 작은 단편으로 분해되어 작은 규모로 샷건 방법을 사용하여 시퀀싱할 수 있다.[16]
BAC 콘티그의 전체 시퀀스는 알려져 있지 않지만 서로 상대적인 방향은 알려져 있다. 이러한 순서를 추론하고 타일링 경로를 구성하는 BAC를 선택하는 몇 가지 방법이 있다. 일반적인 전략은 클론의 위치를 서로 상대적으로 식별한 다음 관심 영역 전체를 덮는 연속 스캐폴드를 형성하는 데 필요한 최소 수의 클론을 선택하는 것이다. 클론의 순서는 클론이 겹치는 방식을 결정하여 추론된다.[17] 겹치는 클론은 시퀀스 태그 부위(STS)를 포함하는 작은 방사성 또는 화학적으로 표지된 프로브를 클론이 인쇄된 마이크로어레이에 혼성화하여 식별할 수 있다.[17] 이러한 방식으로 유전체의 특정 시퀀스를 포함하는 모든 클론이 식별된다. 이러한 클론 중 하나의 끝을 시퀀싱하여 새로운 프로브를 생성하고 염색체 워킹이라는 방법으로 이 과정을 반복할 수 있다.
또는 BAC 라이브러리를 제한 효소로 절단할 수 있다. 여러 단편 크기를 공통적으로 갖는 두 개의 클론은 여러 유사하게 간격을 둔 제한 부위를 공통적으로 포함하기 때문에 겹치는 것으로 추론된다.[17] 이 유전체 매핑 방법은 각 클론에 포함된 일련의 제한 부위를 식별하기 때문에 제한 또는 BAC 지문 분석이라고 한다. 클론 간의 중첩이 발견되고 유전체에 대한 상대적인 순서가 알려지면 전체 유전체를 덮는 이러한 콘티그의 최소 하위 집합의 스캐폴드가 샷건 시퀀싱된다.[15]
계층적 샷건 시퀀싱은 먼저 유전체의 낮은 해상도 지도를 생성하는 작업이 포함되므로 전체 유전체 샷건 시퀀싱보다 느리지만, 전체 유전체 샷건 시퀀싱보다 컴퓨터 알고리즘에 덜 의존한다. 그러나 광범위한 BAC 라이브러리 생성 및 타일링 경로 선택 과정은 계층적 샷건 시퀀싱을 느리고 노동 집약적으로 만든다.
위 예시에서 리드 중 어느 것도 원래 염기 서열의 전체 길이를 포함하지 않지만, 네 개의 리드는 끝부분의 중첩을 사용하여 정렬 및 순서를 정하여 원래 염기 서열로 조립할 수 있다. 실제 이 과정은 모호함과 시퀀싱 오류가 가득한 엄청난 양의 정보를 사용한다. 복잡한 게놈 조립은 반복 서열이 매우 풍부하여 더욱 복잡하며, 이는 유사한 짧은 리드가 완전히 다른 부분에서 나올 수 있음을 의미한다.[3]
이러한 어려움을 극복하고 정확하게 시퀀스를 조립하려면 원래 DNA의 각 부분에 대해 많은 중첩 리드가 필요하다. 예를 들어, 인간 게놈 프로젝트를 완료하기 위해 인간 게놈 대부분은 12X 이상 ''커버리지''로 시퀀싱되었다. 즉, 최종 시퀀스의 각 염기는 평균 12개의 다른 리드에 존재했다. 그럼에도 불구하고, 현재 방법으로는 2004년 기준 약 1%의 유크로마틴 인간 게놈에 대한 신뢰할 수 있는 시퀀스를 분리하거나 조립하는 데 실패했다.[3]
원래 염기 서열은 시퀀스 어셈블리 소프트웨어를 사용하여 읽기 자료로부터 재구성된다. 먼저, 겹치는 읽기 자료들은 컨티그로 알려진 더 긴 합성 서열로 묶인다. 컨티그들은 메이트 페어 간 연결을 따라 스캐폴드로 연결될 수 있다. 라이브러리의 평균 단편 길이를 알고 있고 편차가 좁은 경우, 메이트 페어 위치로부터 컨티그 간 거리를 추론할 수 있다. 컨티그 간 갭 크기에 따라 갭 내 서열을 찾기 위해 서로 다른 기술을 사용할 수 있다. 갭이 작은 경우(5-20kb)에는 중합효소 연쇄 반응(PCR)을 사용하여 해당 영역을 증폭시킨 다음 시퀀싱이 필요하다. 갭이 큰 경우(>20kb)에는 큰 단편을 세균 인공 염색체(BAC)와 같은 특수 벡터에 클론하고, 이어서 벡터를 시퀀싱한다.
3. 4. 커버리지 (Coverage)
커버리지(판독 깊이 또는 깊이)는 재구성된 서열에서 주어진 뉴클레오타이드를 나타내는 평균 판독 횟수이다. 커버리지는 원래 게놈의 길이(''G''), 판독 횟수(''N''), 평균 판독 길이(''L'')를 사용하여 NXL/G 로 계산할 수 있다. 예를 들어, 평균 길이가 500개의 뉴클레오타이드인 8개의 판독값으로 재구성된 2,000개의 염기쌍을 가진 가상 게놈은 2배의 중복성을 갖는다. 이 매개변수를 사용하면 판독값으로 덮인 게놈의 백분율(커버리지라고도 함)과 같은 다른 양을 추정할 수도 있다. 샷건 시퀀싱에서 높은 커버리지는 염기 호출 및 어셈블리의 오류를 극복할 수 있기 때문에 바람직하다. DNA 시퀀싱 이론은 이러한 양의 관계를 다룬다.[3]
때때로 '시퀀스 커버리지'와 '물리적 커버리지'가 구별된다. 시퀀스 커버리지는 염기가 판독된 평균 횟수이다(위에 설명된 대로). 물리적 커버리지는 메이트 페어 판독값으로 염기가 판독되거나 덮이는 평균 횟수이다.[12]
커버리지의 예시는 다음과 같다.
가닥
염기 서열
원본
AGCATGCTGCAGTCATGCTTAGGCTA
첫 번째 샷건 시퀀스
AGCATGCTGCAGTCATGCT-------
두 번째 샷건 시퀀스
AGCATG--------------------
재구성
AGCATGCTGCAGTCATGCTTAGGCTA
위의 예시에서, 리드 중 어느 것도 원래 염기 서열의 전체 길이를 커버하지 않지만, 네 개의 리드는 끝부분의 중첩을 사용하여 정렬하고 순서를 정하여 원래 염기 서열로 조립할 수 있다. 실제로는 이 과정은 모호함과 시퀀싱 오류가 가득한 엄청난 양의 정보를 사용한다. 복잡한 게놈의 조립은 반복 서열이 매우 풍부하기 때문에 더욱 복잡해지며, 이는 유사한 짧은 리드가 완전히 다른 부분에서 나올 수 있음을 의미한다.
이러한 어려움을 극복하고 정확하게 시퀀스를 조립하기 위해서는 원래 DNA의 각 세그먼트에 대해 많은 중첩 리드가 필요하다. 예를 들어, 인간 게놈 프로젝트를 완료하기 위해, 인간 게놈의 대부분은 12X 또는 그 이상의 '커버리지'로 시퀀싱되었다. 즉, 최종 시퀀스의 각 염기는 평균 12개의 다른 리드에 존재했다. 그럼에도 불구하고, 현재 방법으로는 2004년 기준으로 약 1%의 ([유크로마틴|유크로마틴]) 인간 게놈에 대한 신뢰할 수 있는 시퀀스를 분리하거나 조립하는 데 실패했다.[3]
4. 장단점
샷건 시퀀싱은 대규모 시퀀서 배열을 사용하여 전체 유전체를 한 번에 시퀀싱할 수 있어, 전통적인 방식보다 전체 과정을 훨씬 더 효율적으로 만든다는 장점이 있다. 그러나 DNA의 큰 영역을 빠르게 시퀀싱할 수는 있지만, 특히 반복 서열이 많은 진핵생물 유전체의 경우 이러한 영역을 올바르게 연결하는 데 어려움이 있다는 단점도 있다. 시퀀스 조립 프로그램이 더욱 정교해지고 컴퓨팅 성능이 저렴해짐에 따라 이러한 제한은 극복될 수 있을 것으로 보인다.[11]
5. 최신 기술 동향
단편 판독 시퀀싱 및 장편 판독 시퀀싱과 같은 다른 시퀀싱 기술이 사용되고 있다.[18]
"차세대" 시퀀싱이라고도 불리는 단편 판독 시퀀싱은 짧은 길이(25–500bp)의 염기서열을 읽지만, 비교적 짧은 시간(하루 정도) 내에 수십만 또는 수백만 개의 염기서열을 생성한다.[18] 이는 높은 커버리지를 제공하지만, 조립 과정에서 훨씬 더 많은 계산이 필요하다. 이러한 기술은 방대한 데이터량과 전체 게놈 시퀀싱에 걸리는 비교적 짧은 시간 덕분에 생어 시퀀싱보다 훨씬 우수하다.[19]
6. 메타게놈 샷건 시퀀싱 (Metagenomic shotgun sequencing)
메타게놈 샷건 시퀀싱은 환경 샘플에서 얻은 모든 DNA를 무작위로 시퀀싱하여, 해당 환경에 존재하는 미생물 군집의 구성과 기능을 분석하는 방법이다. 400~500 염기쌍 길이의 리드를 갖는 것은, 예를 들어 k-mer 기반 분류 분류기 소프트웨어를 사용하여, DNA가 유래한 생물의 게놈이 이미 알려진 경우 종 또는 균주를 결정하기에 충분하다. 환경 샘플의 차세대 염기 서열 분석을 통해 수백만 개의 리드를 얻으면 장내 미생물군과 같이 수천 종의 복잡한 미생물군에 대한 완전한 개요를 얻을 수 있다. 이는 다음과 같은 장점들을 갖는다.[20]
16S rRNA 증폭체 염기 서열 분석과 비교했을 때, 박테리아에 국한되지 않는다.
증폭체 염기 서열 분석이 속만을 얻는 반면, 균주 수준의 분류가 가능하다.
전체 유전자를 추출하여 메타게놈의 일부로 기능을 지정할 수 있다.
메타게놈 염기 서열 분석의 민감성은 임상적 사용에 매력적인 선택이 되게 한다.[21] 그러나 샘플 또는 염기 서열 분석 파이프라인의 오염 문제를 강조한다.[22]
7. 한국에서의 활용 및 전망
한국에서는 샷건 시퀀싱 기술을 활용하여 질병 진단, 맞춤 의학, 신약 개발, 농작물 품종 개량 등 다양한 분야에서 연구가 활발하게 진행되고 있다. 특히, 더불어민주당은 유전체 정보 기반의 정밀의료 실현을 주요 정책으로 추진하고 있으며, 샷건 시퀀싱 기술은 이러한 정책 목표 달성에 핵심적인 역할을 할 것으로 기대된다.
[1]
논문
A strategy of DNA sequencing employing computer programs
1979
[2]
논문
Shotgun DNA sequencing using cloned DNase I-generated fragments
1981
[3]
논문
Finishing the euchromatic sequence of the human genome
2004-10-21
[4]
논문
The complete nucleotide sequence of an infectious clone of cauliflower mosaic virus by M13mp7 shotgun sequencing
1981-06-25
[5]
논문
Profile of Joachim Messing
2016-07-19
[6]
논문
Closure strategies for random DNA sequencing
1991-08
[7]
논문
Automated DNA sequencing of the human HPRT locus
1990-04
[8]
논문
Pairwise end sequencing: a unified approach to genomic mapping and sequencing
1995-03
[9]
논문
Whole-genome random sequencing and assembly of Haemophilus influenzae Rd
1995
[10]
논문
The genome sequence of Drosophila melanogaster
http://faculty.evans[...]
2017-10-25
[11]
논문
Bioinformatics challenges of new sequencing technology
2008-03
[12]
논문
Advances in understanding cancer genomes through second-generation sequencing
[13]
논문
Genome Sequencing
2005-09-09
[14]
논문
Shotgunning the Human Genome: A Personal View
2005-09-09
[15]
서적
A Primer of Genome Science
[16]
논문
A Graph-Theoretical Approach to the Selection of the Minimum Tiling Path from a Physical Map
2013-03
[17]
논문
Genome Mapping
2005-09-09
[18]
논문
Next-Generation Sequencing: From Basic Research to Diagnostics
2009-04-01
[19]
논문
Sequencing technologies — the next generation
2010-01
[20]
논문
A Review of Bioinformatics Tools for Bio-Prospecting from Metagenomic Sequence Data
2017-03-06
[21]
논문
Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection
2019-01-24
[22]
논문
Impact of Contaminating DNA in Whole-Genome Amplification Kits Used for Metagenomic Shotgun Sequencing for Infection Diagnosis
2017-06
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.