서열 정렬
"오늘의AI위키"의 AI를 통해 더욱 풍부하고 폭넓은 지식 경험을 누리세요.
1. 개요
서열 정렬은 두 개 이상의 서열을 비교하여 유사성을 파악하는 생물정보학 기법이다. 불일치는 점 돌연변이, 갭은 인델로 해석하며, 단백질 서열 정렬에서는 아미노산 유사성이 보존 정도를 나타낸다. 정렬 방법에는 전역 정렬과 지역 정렬이 있으며, 동적 프로그래밍, 휴리스틱 알고리즘 등 다양한 계산적 접근 방식이 사용된다. 서열 정렬은 그래픽 및 텍스트 형식으로 표현되며, 쌍별 정렬, 다중 서열 정렬, 구조 정렬 등 다양한 유형이 존재한다. 이는 계통 발생 분석, 유전자 위치 파악, 게놈 조립 등 다양한 생물학적 응용 분야에서 활용되며, 자연어 처리, 사회 과학 등 비생물학적 분야에서도 사용된다. 서열 정렬 소프트웨어는 다양한 알고리즘과 정렬 유형을 지원하며, BAliBASE와 같은 벤치마크를 통해 성능을 평가한다.
더 읽어볼만한 페이지
- 생물정보학 알고리즘 - BLAST (생명공학기술)
BLAST는 DNA 또는 단백질 시퀀스 간의 유사성을 빠르게 찾기 위해 사용되는 생물정보학 프로그램으로, 대규모 데이터베이스에서 시퀀스 정렬을 효율적으로 수행하며 다양한 생물학적 연구에 활용된다. - 진화발생생물학 - 전사인자
전사 인자는 DNA 특정 서열에 결합하여 유전자 발현을 조절하는 단백질로서, RNA 중합효소와 함께 전사를 조절하여 유전 정보 전달에 핵심적인 역할을 하며, 발생, 신호 전달, 환경 반응, 세포 주기 조절 등 다양한 생물학적 과정에 관여한다. - 진화발생생물학 - 유전자 발현의 조절
유전자 발현의 조절은 세포 내에서 특정 유전자의 발현 시기와 양을 제어하는 복잡한 과정으로, 신호 전달부터 전사, 번역 후 변형에 이르기까지 다양한 단계에서 DNA 접근성 조절, 전사 조절, RNA 수송, 번역 조절, mRNA 분해 등 다양한 메커니즘을 포함하며, 후생유전학적 조절은 DNA 서열 변화 없이 유전자 발현에 영향을 미친다. - 생물정보학 - Rosetta@home
Rosetta@home은 분산 컴퓨팅 플랫폼 BOINC를 활용하여 단백질 구조 예측 연구를 수행하며, 신약 개발 및 질병 연구에 기여하는 것을 목표로 한다. - 생물정보학 - 발현체학
| 서열 정렬 | |
|---|---|
| 개요 | |
| 유형 | 생물 정보학 |
| 정의 | DNA, RNA, 또는 단백질 서열과 같은 생물학적 서열을 배열하는 방법 |
| 목적 | |
| 1차 목적 | 서열 간의 유사성, 또는 상동성을 식별하고 서열의 기능적, 구조적, 진화적 관계를 추론 |
| 추가 목적 | 진화 연구 약물 개발 단백질 구조 예측 |
| 종류 | |
| 글로벌 서열 정렬 (Global alignment) | 전체 서열을 정렬 |
| 로컬 서열 정렬 (Local alignment) | 서열 내에서 유사한 영역을 식별 |
| 다중 서열 정렬 (Multiple sequence alignment) | 셋 이상의 서열을 정렬 |
| 쌍별 서열 정렬 (Pairwise sequence alignment) | 두 서열을 정렬 |
| 방법 | |
| 점 행렬 방법 (Dot matrix methods) | 서열 유사성을 시각적으로 표현 |
| 동적 프로그래밍 (Dynamic programming) | 최적의 정렬을 보장하는 알고리즘 |
| 단어 방법 (Word methods) | 짧은 서열을 기반으로 빠른 검색 |
| 숨겨진 마르코프 모델 (Hidden Markov models) | 서열 프로필을 사용하여 정렬 |
| 알고리즘 | |
| Needleman-Wunsch 알고리즘 | 글로벌 서열 정렬에 사용 |
| Smith-Waterman 알고리즘 | 로컬 서열 정렬에 사용 |
| BLAST | 서열 유사성 검색에 사용 |
| FASTA | 서열 유사성 검색에 사용 |
| ClustalW | 다중 서열 정렬에 사용 |
| MUSCLE | 다중 서열 정렬에 사용 |
| MAFFT | 다중 서열 정렬에 사용 |
| 응용 | |
| 유전자 예측 | 유전자 위치 식별 |
| 계통 발생 | 종 간의 진화적 관계 연구 |
| 기능적 모티프 식별 | 단백질 기능 예측 |
| 구조적 모티프 식별 | 단백질 구조 예측 |
| 상동성 모델링 | 단백질 3차원 구조 예측 |
| 소프트웨어 도구 | |
| BLAST | NCBI BLAST |
| ClustalW | ClustalW |
| MUSCLE | MUSCLE |
| MAFFT | MAFFT |
| T-Coffee | T-Coffee |
| 일반적인 기호 | |
| "*" (별표) | 완전히 동일한 잔기 |
| ":" (콜론) | 높은 점수의 유사한 잔기 |
| "." (점) | 낮은 점수의 유사한 잔기 |
2. 해석
두 정렬된 서열이 공통 조상을 공유하는 경우, 불일치는 점 돌연변이로, 갭은 인델(삽입 또는 삭제 돌연변이)로 해석될 수 있으며, 이는 두 계통 중 하나 이상에서 서로 분기된 이후 시간에 도입된 것이다.[3] 단백질의 서열 정렬에서, 서열의 특정 위치를 차지하는 아미노산 간의 유사성 정도는 특정 영역 또는 서열 모티프가 계통 간에 얼마나 보존되었는지에 대한 대략적인 척도로 해석될 수 있다. 서열의 특정 영역에서 치환이 없거나 매우 보수적인 치환(즉, 측쇄가 유사한 생화학적 특성을 갖는 아미노산의 치환)만 있는 것은 이 영역이 구조적 또는 기능적으로 중요하다는 것을 시사한다.
서열 정렬에 대한 계산적 접근 방식은 일반적으로 전역 정렬과 지역 정렬 두 가지로 나뉜다. 전역 정렬은 모든 질의 서열의 전체 길이에 걸쳐 정렬을 수행하는 전역 최적화의 한 형태이다. 반면, 지역 정렬은 전체적으로 매우 다른 긴 서열 내에서 유사한 영역을 식별한다. 지역 정렬이 선호되기도 하지만, 유사한 영역을 식별하기가 더 어려워 계산하기 더 까다로울 수 있다.[4]
3. 정렬 방법
서열 정렬 문제에는 동적 프로그래밍처럼 느리지만 형식적으로 정확한 방법과, 대규모 데이터베이스 검색을 위해 설계되었으며 최상의 일치를 보장하지는 않는 효율적인 휴리스틱 알고리즘 또는 확률적 방법 등 다양한 계산 알고리즘이 적용된다.
3. 1. 전역 정렬과 지역 정렬
전역 정렬은 각 서열의 모든 잔기를 정렬하려는 시도로, 질의 집합의 서열이 유사하고 대략 같은 크기일 때 가장 유용하다.[4] 일반적인 전체 정렬 기술은 니들만-분쉬 알고리즘이며, 이는 동적 프로그래밍을 기반으로 한다. 국소 정렬은 더 큰 서열 맥락 내에서 유사한 영역 또는 유사한 서열 모티프를 포함할 것으로 의심되는, 유사하지 않은 서열에 더 유용하다.[4] 스미스-워터만 알고리즘은 동일한 동적 프로그래밍 방식을 기반으로 하지만 시작 및 종료 지점을 임의로 선택할 수 있는 일반적인 국소 정렬 방법이다.
반-전체 또는 "글로컬"('''.'''glo'''bal-lo'''cal'''의 줄임말) 방법은 두 서열의 최적의 부분 정렬을 찾는다.[4] 이는 한 서열의 다운스트림 부분이 다른 서열의 업스트림 부분과 겹칠 때 특히 유용하다. 이 경우 전체 정렬과 국소 정렬 모두 완전히 적절하지 않다. 전체 정렬은 겹치는 영역을 넘어 정렬을 확장하려고 시도하는 반면 국소 정렬은 겹치는 영역을 완전히 커버하지 못할 수 있다. 반-전체 정렬이 유용한 또 다른 경우는 한 서열이 짧고(예: 유전자 서열) 다른 서열이 매우 긴 경우(예: 염색체 서열)이다. 이 경우 짧은 서열은 전체적으로(완전하게) 정렬되어야 하지만 긴 서열에 대해서는 국소적인(부분적인) 정렬만 원한다.
글로벌 정렬은 배열 내의 모든 잔기가 정렬되도록 하는 방법으로, 거의 동일한 길이를 가진 배열 간의 비교에 유효하다. 로컬 정렬은 배열이 전체적으로는 유사하지 않지만 부분적인 유사성을 찾고 싶을 때 유효하다.
4. 표현
정렬은 일반적으로 그래픽 형식과 텍스트 형식으로 모두 표현된다. 거의 모든 서열 정렬 표현에서, 정렬된 잔기는 연속적인 열에 나타나도록 배열된 행으로 서열을 작성한다. 텍스트 형식에서, 동일하거나 유사한 문자를 포함하는 정렬된 열은 보존 기호 시스템으로 표시된다. 별표(*) 또는 파이프(|) 기호는 두 열 간의 동일성을 나타내는 데 사용된다. 보존적 치환에는 콜론(:), 반보존적 치환에는 마침표(.)가 사용되기도 한다. 많은 서열 시각화 프로그램은 개별 서열 요소의 속성에 대한 정보를 표시하기 위해 색상을 사용하기도 한다. DNA 및 RNA 서열에서 이는 각 뉴클레오티드에 자체 색상을 할당하는 것과 같다. 단백질 정렬에서 색상은 주어진 아미노산 치환의 보존을 판단하는 데 도움이 되도록 아미노산 속성을 나타내는 데 자주 사용된다. 다중 서열의 경우 각 열의 마지막 행은 컨센서스 서열인 경우가 많다. 컨센서스 서열은 각 뉴클레오티드 또는 아미노산 문자의 크기가 보존 정도에 해당하는 서열 로고와 함께 그래픽 형식으로 표현되는 경우가 많다.[5]
서열 정렬은 다양한 텍스트 기반 파일 형식으로 저장할 수 있으며, 그 중 상당수는 원래 특정 정렬 프로그램 또는 구현과 함께 개발되었다. 대부분의 웹 기반 도구는 FASTA 형식 및 GenBank 형식과 같은 제한된 수의 입력 및 출력 형식을 허용하며, 출력은 쉽게 편집할 수 없다.
5. 쌍별 정렬
쌍별 서열 정렬은 두 질의 서열 간의 최적 일치 분할(지역적 또는 전역적) 정렬을 찾는 방법이다. 계산 효율이 높아 극도의 정밀도가 필요하지 않은 경우(예: 데이터베이스에서 질의와 유사성이 높은 서열 검색)에 자주 사용된다.[1] 쌍별 정렬은 한 번에 두 개의 서열 사이에서만 사용할 수 있다.
쌍별 정렬을 생성하는 주요 방법에는 점 행렬 방법, 동적 프로그래밍, 단어 방법이 있다.[1] 다중 서열 정렬 기술을 사용하여 서열 쌍을 정렬할 수도 있다. 각 방법은 고유한 강점과 약점을 가지지만, 세 가지 쌍별 방법 모두 정보 내용이 낮은 매우 반복적인 서열(특히 두 서열에서 반복 횟수가 다른 경우)에 어려움을 겪는다.
쌍별 시퀀스 정렬은 두 서열 간의 정렬로, 부분적 또는 전체적인 유사성을 자세히 조사할 때 사용한다.
5. 1. 최대 고유 일치 (MUM)
최대 고유 일치(MUM)는 주어진 쌍별 정렬의 유용성을 정량화하는 한 가지 방법으로, 두 쿼리 시퀀스 모두에 나타나는 가장 긴 서브시퀀스이다. 일반적으로 더 긴 MUM 서열은 더 가까운 관련성을 나타낸다.[8] 전산 생물학에서 유전체의 다중 서열 정렬에서 MUM을 식별하는 것은 MUMmer와 같은 더 큰 정렬 시스템의 첫 번째 단계이다. 앵커는 두 유전체 사이의 영역으로, 매우 유사하다. MUM이 무엇인지 이해하기 위해 약어의 각 단어를 분석할 수 있다. 일치는 정렬할 두 시퀀스 모두에 서브스트링이 나타남을 의미한다. 고유는 서브스트링이 각 시퀀스에서 한 번만 나타남을 의미한다. 마지막으로, 최대는 서브스트링이 이전 두 가지 요구 사항을 모두 충족하는 더 큰 다른 문자열의 일부가 아님을 나타낸다. 이 아이디어는 정확히 일치하고 각 유전체에서 한 번만 나타나는 긴 시퀀스는 거의 확실히 전역 정렬의 일부라는 것이다.더 정확하게는 다음과 같다.
> 두 유전체 A와 B가 주어졌을 때, 최대 고유 일치(MUM) 서브스트링은 지정된 최소 길이 d(기본적으로 d=20)보다 긴 A와 B의 공통 서브스트링으로,
> * 최대 길이, 즉 불일치를 발생시키지 않고 양쪽 끝에서 확장할 수 없다. 그리고
> * 두 시퀀스 모두에서 고유하다.[9]
5. 2. 점 행렬 방법
점 행렬 방식은 개별 시퀀스 영역에 대한 일련의 정렬을 암묵적으로 생성하며, 정성적이고 개념적으로 단순하지만 대규모로 분석하는 데 시간이 오래 걸린다. 노이즈가 없는 경우 점 행렬 플롯에서 삽입, 삭제, 반복 또는 역반복과 같은 특정 시퀀스 특징을 시각적으로 쉽게 식별할 수 있다. 점 행렬 플롯을 구성하기 위해 두 시퀀스는 2차원 행렬의 맨 위 행과 맨 왼쪽 열에 기록되고 적절한 열의 문자가 일치하는 지점에 점이 찍힌다. 이는 전형적인 재귀 플롯이다. 일부 구현에서는 두 문자의 유사성 정도에 따라 점의 크기나 강도를 변경하여 보존적 치환을 수용한다. 매우 밀접하게 관련된 시퀀스의 점 플롯은 행렬의 주 대각선을 따라 단일 선으로 나타난다.
도트 매트릭스 방식은 결과적으로 각 서열 내의 영역 간 정렬, 즉 다수의 정렬 결과를 얻는다. 이 방식은 간단하지만, 대규모 계산 시 시간이 소요된다. 다만, 노이즈만 없다면 서열 간의 상동성 측면에서 특징적인 영역을 육안으로 쉽게 식별할 수 있다. 예를 들어 삽입, 결실, 반복 서열, Inverted repeat 등은 2차원 도트 매트릭스 플롯에서 찾을 수 있다.
도트 매트릭스 플롯을 작성하려면 먼저 2차원 행렬의 행에 한 개의 서열을 할당하고, 열에 다른 서열을 할당한다. 그리고 속성이 일치하는 행과 열이 교차하는 지점에 점을 그린다. 도트 플롯의 구현에 따라 유사성의 정도를 점의 크기나 농도로 나타내기도 한다.
5. 3. 동적 프로그래밍
동적 계획법 기술은 니들만-분쉬 알고리즘을 통해 전체 정렬을 생성하고, 스미스-워터만 알고리즘을 통해 지역 정렬을 생성하는 데 적용될 수 있다. 일반적인 단백질 정렬은 치환 행렬을 사용하여 아미노산 일치 또는 불일치에 점수를 할당하고, 한 서열의 아미노산을 다른 서열의 갭에 일치시키는 데 갭 패널티를 할당한다.[4] DNA와 RNA 정렬은 점수 행렬을 사용할 수 있지만, 실제로는 단순히 양의 일치 점수, 음의 불일치 점수, 음의 갭 패널티를 할당하는 경우가 많다.표준 선형 갭 비용에 대한 일반적인 확장에는 아핀 갭 비용이 있으며, 갭을 여는 것과 갭을 확장하는 것에 대해 두 가지 다른 갭 패널티가 적용된다. 일반적으로 갭을 여는 패널티가 갭을 확장하는 패널티보다 훨씬 크다(예: 갭 열기에 -10, 갭 확장에 -2).[10][11]
5. 4. 단어 방법
단어 방법(*k*-튜플 방법)은 최적의 정렬 해답을 찾는 것을 보장하지는 않지만 동적 프로그래밍보다 훨씬 효율적인 휴리스틱 방법이다.[1] 이러한 방법은 후보 서열의 상당 부분이 쿼리 서열과 본질적으로 유의미한 일치를 보이지 않는다는 것을 알고 있는 대규모 데이터베이스 검색에 특히 유용하다. 단어 방법은 데이터베이스 검색 도구인 FASTA와 BLAST 계열에서 구현된 것으로 가장 잘 알려져 있다.[1]단어 방법은 쿼리 서열에서 일련의 짧고 겹치지 않는 부분 서열("단어")을 식별한 다음 후보 데이터베이스 서열과 일치시킨다. 비교되는 두 서열에서 단어의 상대 위치를 빼서 오프셋을 얻는다. 여러 개의 서로 다른 단어가 동일한 오프셋을 생성하는 경우 이것은 정렬 영역을 나타낸다. 이러한 영역이 감지된 경우에만 이러한 방법은 더 민감한 정렬 기준을 적용한다. 따라서 상당한 유사성이 없는 서열과의 불필요한 비교가 많이 제거된다.
FASTA 방법에서 사용자는 데이터베이스를 검색하는 데 사용할 단어 길이로 값 ''k''를 정의한다. 이 방법은 더 느리지만 ''k'' 값이 낮을수록 더 민감하며, 매우 짧은 쿼리 서열을 포함하는 검색에도 선호된다. BLAST 계열의 검색 방법은 먼 관계의 서열 일치 검색과 같은 특정 유형의 쿼리에 최적화된 여러 알고리즘을 제공한다. BLAST는 정확성을 크게 저해하지 않으면서 FASTA의 더 빠른 대안을 제공하기 위해 개발되었다. FASTA와 마찬가지로 BLAST는 길이 ''k''의 단어 검색을 사용하지만 FASTA와 같이 모든 단어 일치가 아닌 가장 유의미한 단어 일치만 평가한다. 대부분의 BLAST 구현은 쿼리 및 데이터베이스 유형에 최적화된 고정된 기본 단어 길이를 사용하며 반복적이거나 매우 짧은 쿼리 서열로 검색하는 경우와 같은 특수한 상황에서만 변경된다. 구현은 [https://www.ncbi.nlm.nih.gov/BLAST/ NCBI BLAST]와 같은 여러 웹 포털을 통해 찾을 수 있다.
6. 다중 서열 정렬
다중 서열 정렬은 셋 이상의 서열을 대상으로 하는 쌍별 서열 정렬의 확장으로, 진화적으로 연관된 서열 간의 보존된 영역을 식별하는 데 유용하다. 다중 정렬은 주어진 모든 서열에 대해 정렬을 시도하며, 진화와 관련된 서열 그룹에서 보존 서열 영역을 결정하는 데 자주 사용된다. 이러한 보존 서열 모티프는 효소의 촉매 활성 부위를 찾는 데 사용되는 구조 및 반응 메커니즘 정보와 결합하여 활용된다. 또한, 정렬은 계통수를 구축하여 진화적 관계를 보여주는 데 사용된다.
다중 서열 정렬은 계산적으로 생성하기 어렵고, 대부분의 문제 공식은 NP-완전인 조합 최적화 문제로 이어진다.[12][13] 그럼에도 불구하고, 생물정보학에서 이러한 정렬의 유용성으로 인해 셋 이상의 서열을 정렬하기 위한 다양한 방법들이 개발되었다.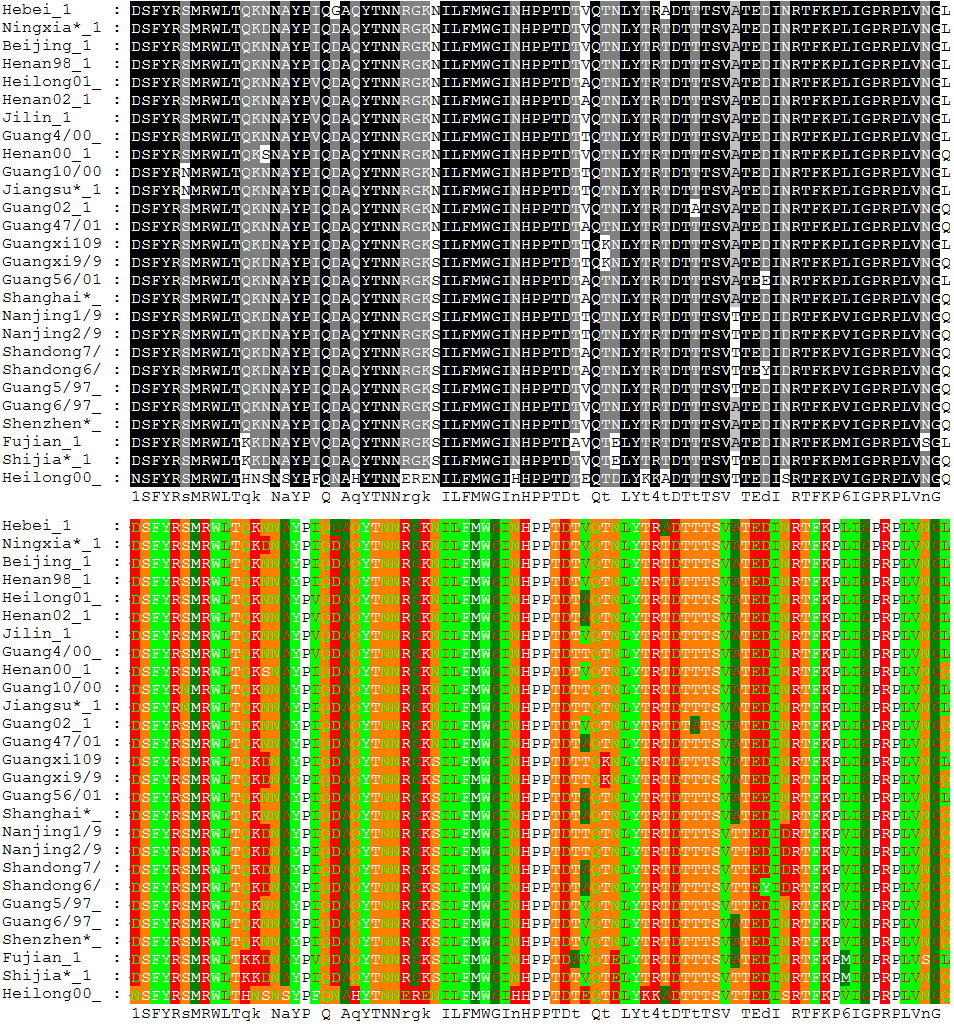
6. 1. 동적 프로그래밍
동적 프로그래밍 기법은 이론적으로는 어떤 수의 시퀀스에도 적용할 수 있지만, 시간과 메모리 측면에서 계산 비용이 많이 들기 때문에, 가장 기본적인 형태로는 세 개 또는 네 개 이상의 시퀀스에는 거의 사용되지 않는다. 이 방법은 쿼리에 있는 시퀀스의 수인 ''n'' 차원의 시퀀스 행렬을 구성해야 하며, 이는 두 시퀀스로 구성된다. 표준 동적 프로그래밍은 먼저 모든 쿼리 시퀀스 쌍에 사용된 다음, 중간 위치에서 가능한 일치 또는 갭을 고려하여 "정렬 공간"을 채워 결국 각 두 시퀀스 정렬 간의 정렬을 구성한다. 이 기법은 계산 비용이 많이 들지만, 전역 최적 솔루션을 보장하므로, 정확하게 정렬해야 하는 시퀀스가 몇 개 없을 때 유용하다.[14] "쌍의 합" 목적 함수에 의존하는 동적 프로그래밍의 계산 요구 사항을 줄이는 한 가지 방법이 [https://www.ncbi.nlm.nih.gov/CBBresearch/Schaffer/msa.html MSA] 소프트웨어 패키지에 구현되었다.[14]6. 2. 점진적 방법
점진적 방법, 계층적 방법 또는 트리 방법은 가장 유사한 서열을 먼저 정렬한 다음, 덜 관련된 서열 또는 그룹을 순차적으로 정렬에 추가하여 다중 서열 정렬을 생성한다. 서열 관련성을 설명하는 초기 트리는 FASTA와 유사한 발견적 쌍별 정렬 방법을 포함할 수 있는 쌍별 비교를 기반으로 한다. 점진적 정렬 결과는 "가장 관련성이 높은" 서열의 선택에 따라 달라지며, 따라서 초기 쌍별 정렬의 부정확성에 민감할 수 있다. 대부분의 점진적 다중 서열 정렬 방법은 쿼리 집합의 서열에 관련성에 따라 가중치를 부여하여 초기 서열의 잘못된 선택 가능성을 줄여 정렬 정확도를 향상시킨다.[12][13]Clustal 점진적 구현[15][16][17]의 많은 변형이 다중 서열 정렬, 계통수 구성, 그리고 단백질 구조 예측의 입력으로 사용된다. 점진적 방법의 느리지만 더 정확한 변형은 T-Coffee로 알려져 있다.[18]
6. 3. 반복적 방법
반복 방법은 초기 쌍별 정렬의 정확성에 대한 과도한 의존성을 개선하려 시도하며, 이는 점진적 방법의 약점이다.[19] 반복 방법은 초기 전역 정렬을 할당한 다음 시퀀스 하위 집합을 재정렬하여 선택된 정렬 점수 매김 방법을 기반으로 하는 목적 함수를 최적화한다. 재정렬된 하위 집합은 다음 반복의 다중 시퀀스 정렬을 생성하기 위해 자체적으로 정렬된다. 시퀀스 하위 그룹 및 목적 함수를 선택하는 다양한 방법이 검토되어 있다.[19]6. 4. 모티프 찾기
모티프 찾기(프로파일 분석이라고도 함)는 질의 집합 내 서열들 사이에서 짧게 보존된 서열 모티프를 정렬하려는 전역 다중 서열 정렬을 구성한다. 이는 일반적으로 먼저 일반적인 전역 다중 서열 정렬을 구성한 후, 고도로 보존된 영역을 분리하여 일련의 프로파일 행렬을 구성함으로써 수행된다. 각 보존 영역에 대한 프로파일 행렬은 점수 행렬과 유사하게 배열되지만, 각 위치에서 각 아미노산 또는 뉴클레오타이드에 대한 빈도 수는 보다 일반적인 경험적 분포가 아닌 보존 영역의 문자 분포에서 파생된다. 그런 다음 프로파일 행렬을 사용하여 다른 서열에서 해당 모티프의 발생을 검색한다. 원래의 데이터 집합에 소수의 서열 또는 고도로 관련된 서열만 포함된 경우, 모티프에 표시된 문자 분포를 정규화하기 위해 가상 계수가 추가된다.[12][13]6. 5. 컴퓨터 과학에서 영감을 받은 기술
은닉 마르코프 모델, 유전자 알고리즘, 시뮬레이티드 어닐링 등 컴퓨터 과학의 다양한 최적화 알고리즘이 다중 시퀀스 정렬 문제에 적용되었다.[20] Burrows–Wheeler 변환은 Bowtie 및 BWA와 같은 도구에서 빠른 짧은 리드 정렬에 성공적으로 적용되었다.7. 구조 정렬
구조 정렬은 단백질이나 RNA 서열 정렬을 할 때 이차 구조 및 삼차 구조 정보를 활용하는 방법이다. 단백질과 RNA 구조는 서열보다 진화 과정에서 더 잘 보존되기 때문에, 구조 정렬은 서열 비교로는 유사성을 파악하기 어려운, 매우 멀리 관련된 서열들 사이에서도 더 신뢰성 있는 결과를 제공한다.[21] 단백질 구조 예측에서 구조 정렬은 정렬 결과를 평가하는 "골드 스탠다드"로 사용된다.[22] 그러나 실제 구조 예측에서는 대상 서열의 구조를 알 수 없으므로 구조 정렬을 직접 적용할 수는 없다. 따라서 구조 예측의 주요 과제는 서열 정보만을 가지고 구조적으로 정확한 정렬을 만들어내는 것이다.[22]
구조 정렬은 주로 단백질의 이차 구조와 삼차 구조 정보를 이용하여 서열 정렬을 수행하지만, RNA에도 적용 가능하다. 단백질의 경우, 삽입과 결손은 대부분 무작위 코일(random coil)이나 루프(loop) 영역에서 발생한다. 따라서 구조 정렬은 서열 정렬 후 이러한 삽입 및 결손 서열을 무작위 코일이나 루프에 위치하도록 재정렬하는 과정을 포함한다.
7. 1. DALI
DALI 방법(거리 행렬 정렬)은 쿼리 시퀀스에서 연속적인 헥사펩타이드 간의 접촉 유사성 패턴을 기반으로 구조 정렬을 구축하는 조각 기반 방법이다.[23] 이는 쌍별 또는 다중 정렬을 생성하고 단백질 데이터 뱅크(PDB)에서 쿼리 시퀀스의 구조적 이웃을 식별할 수 있다. 이 방법은 FSSP 구조 정렬 데이터베이스(단백질의 구조-구조 정렬을 기반으로 한 폴드 분류, 즉 구조적으로 유사한 단백질 패밀리)를 구축하는 데 사용되었다.7. 2. SSAP
SSAP(순차 구조 정렬 프로그램)는 구조 공간에서 원자 대 원자 벡터를 비교점으로 사용하는 동적 프로그래밍 기반의 구조 정렬 방법이다. 최초 설명 이후 다중 정렬 및 쌍별 정렬을 포함하도록 확장되었으며,[24] CATH(Class, Architecture, Topology, Homology) 단백질 폴드 계층적 데이터베이스 분류의 구축에 사용되었다.[25] CATH 데이터베이스는 [http://www.cathdb.info/ CATH Protein Structure Classification]에서 접속할 수 있다.7. 3. 조합 확장
구조 정렬의 조합 확장 방법은 국소 기하학을 사용하여 분석 중인 두 단백질의 짧은 조각을 정렬한 다음, 이러한 조각들을 더 큰 정렬로 조립하여 쌍으로 된 구조 정렬을 생성한다.[26] 강체 평균 제곱근 거리, 잔기 거리, 국소 2차 구조, 잔기 이웃 소수성과 같은 주변 환경적 특징과 같은 측정값을 기반으로 "정렬된 단편 쌍"이라고 하는 국소 정렬이 생성된다. 이 국소 정렬은 미리 정의된 컷오프 기준 내에서 가능한 모든 구조 정렬을 나타내는 유사성 행렬을 구축하는 데 사용된다. 그런 다음 성장하는 정렬을 한 번에 한 조각씩 확장하여 하나의 단백질 구조 상태에서 다른 단백질 구조 상태로의 경로가 행렬을 통해 추적된다. 최적의 경로는 조합 확장 정렬을 정의한다. 이 방법을 구현하고 단백질 데이터 뱅크의 구조에 대한 쌍으로 된 정렬 데이터베이스를 제공하는 웹 기반 서버는 https://web.archive.org/web/19981203071023/http://cl.sdsc.edu/ 조합 확장 웹사이트에 있다.8. 계통 발생 분석
계통발생학은 서열 정렬을 광범위하게 사용하여 계통수를 구축하고 해석한다. 쿼리 세트의 서열이 서로 얼마나 다른지는 서열의 진화적 거리와 질적으로 관련이 있다.[27] 높은 서열 동일성은 해당 서열이 비교적 젊은 가장 최근 공통 조상을 가지고 있음을 시사하고, 낮은 동일성은 분기가 더 오래되었음을 시사한다.
점진적 다중 정렬 기술은 관련 순서대로 서열을 정렬에 통합하기 때문에 필연적으로 계통수를 생성한다. 다중 서열 정렬 및 계통수를 조합하는 다른 기술은 먼저 트리를 평가하고 정렬한 다음 최고 점수 트리를 기반으로 다중 서열 정렬을 계산한다. 계통수 구축에 일반적으로 사용되는 방법은 주로 휴리스틱인데, 최적의 트리 선택 문제, 즉 최적의 다중 서열 정렬 선택 문제는 NP-hard이기 때문이다.[28]
8. 1. 유의성 평가
서열 정렬은 생물정보학에서 서열 유사성을 식별하고, 계통수를 생성하는 데 유용하지만, 그 생물학적 관련성이 항상 명확한 것은 아니다. 정렬이 공통 조상으로부터 유래한 서열 간의 진화적 변화 정도를 반영한다고 가정하지만, 수렴 진화로 인해 진화적으로 관련이 없는 단백질 간에도 유사성이 나타날 수 있기 때문이다.[27]BLAST와 같은 데이터베이스 검색에서 통계적 방법을 사용하여 특정 정렬이 우연히 발생할 가능성을 판단할 수 있다. 이 가능성은 데이터베이스 크기, 구성, 반복 서열 등에 따라 달라진다. 특히, 데이터베이스가 쿼리 서열과 동일한 유기체의 서열로만 구성된 경우, 우연히 정렬을 찾을 가능성이 높아진다. BLAST는 이러한 통계적 왜곡을 피하기 위해 쿼리에서 반복 서열을 자동으로 필터링한다.[27]
8. 2. 점수 함수
서열 정렬에서 좋은 결과를 얻으려면 알려진 서열에 대한 생물학적 또는 통계적 관찰을 반영하는 스코어링 함수를 선택하는 것이 중요하다. 단백질 서열은 주어진 문자 대 문자 치환의 확률을 반영하는 치환 행렬을 사용하여 자주 정렬된다. 마가렛 데이호프가 정의한 PAM 행렬(Point Accepted Mutation matrices)은 특정 아미노산 돌연변이의 속도와 확률에 대한 진화적 근사를 명시적으로 인코딩한다. 또 다른 일반적인 스코어링 행렬인 BLOSUM(Blocks Substitution Matrix)은 경험적으로 유도된 치환 확률을 인코딩한다. 두 유형의 행렬 변형은 서로 다른 수준의 발산을 가진 서열을 감지하는 데 사용되므로, BLAST 또는 FASTA 사용자는 검색을 더 밀접하게 관련된 일치 항목으로 제한하거나 더 발산된 서열을 감지하도록 확장할 수 있다. 갭 페널티는 뉴클레오티드 및 단백질 서열 모두에서 갭(진화 모델에서 삽입 또는 삭제 돌연변이)의 도입을 설명하므로, 페널티 값은 이러한 돌연변이의 예상 속도에 비례해야 한다.[3] 따라서 생성된 정렬의 품질은 스코어링 함수의 품질에 따라 달라진다.9. 기타 생물학적 응용
염기 서열 분석된 RNA(예: 발현 서열 태그 및 전체 길이 mRNA)는 유전자 위치를 파악하고 대립 유전자 접합[37] 및 RNA 편집[38]에 대한 정보를 얻기 위해 염기 서열 분석된 게놈에 정렬될 수 있다. 염기 서열 정렬은 염기 서열이 겹치는 부분을 찾아 ''콘티그''(긴 염기 서열)를 형성하는 게놈 조립의 일부이다.[39] 또 다른 용도는 SNP 분석으로, 서로 다른 개체로부터 얻은 염기 서열을 정렬하여 집단에서 자주 다른 단일 염기쌍을 찾는 데 사용된다.[40] 선택적 스플라이싱에도 사용된다.
10. 비생물학적 응용
자연어 처리와 사회 과학 분야에서 서열 정렬 방법이 응용되고 있으며, 니들만-분시 알고리즘은 일반적으로 최적 매칭이라고 불린다.[41] 자연어 생성 알고리즘에서 단어를 선택할 요소 집합을 생성하는 기술은 생물 정보학의 다중 서열 정렬 기술을 차용하여 자동 정리 증명의 언어학적 버전을 생성했다.[42] 역사 및 비교 언어학 분야에서 서열 정렬은 언어학자들이 전통적으로 언어를 재구성하는 비교 언어학적 방법을 부분적으로 자동화하는 데 사용되었다.[43] 또한, 비즈니스 및 마케팅 연구에서도 시간에 따른 구매 일련의 분석에 다중 서열 정렬 기술을 적용했다.[44]
11. 소프트웨어
서열 정렬에 사용되는 일반적인 소프트웨어 도구에는 ClustalW2[45], T-coffee[46]가 있으며, 데이터베이스 검색에는 BLAST[47], FASTA3x[48]가 사용된다. DNASTAR Lasergene, Geneious, PatternHunter와 같은 상용 도구도 이용 가능하다. [https://bio.tools/?page=1&function=%22Sequence%20alignment%22&sort=score bio.tools] 레지스트리에는 [http://edamontology.org/operation_0292 서열 정렬]을 수행하는 도구들이 나열되어 있다.
BAliBASE[49]는 서열 정렬 알고리즘과 소프트웨어를 비교하는 데 사용되는 표준화된 벤치마크 참조 다중 서열 정렬 세트이다. BAliBASE는 구조적 정렬로 구성되어 순수한 서열 기반 방법의 표준으로 간주될 수 있다. 일반적인 정렬 문제에 대한 여러 정렬 방법의 상대적 성능이 표로 정리되어 BAliBASE에 온라인으로 게시되어 있다.[50][51] 단백질 워크벤치 STRAP 내에서 12가지 정렬 도구에 대한 BAliBASE 점수 목록을 확인할 수 있다.[52]
참조
[1]
서적
Bioinformatics: Sequence and Genome Analysis
Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY.
[2]
웹사이트
Clustal FAQ #Symbols
http://www.ebi.ac.uk[...]
2014-12-08
[3]
논문
Predicting deleterious amino acid substitutions
2001-05
[4]
논문
Comparative analysis of the quality of a global algorithm and a local algorithm for alignment of two sequences
[5]
논문
Sequence logos: a new way to display consensus sequences
[6]
웹사이트
Sequence Alignment/Map Format Specification
https://samtools.git[...]
[7]
논문
Glocal alignment: finding rearrangements during alignment
[8]
논문
Alignment of whole genomes
1999
[9]
서적
Algorithms in Bioinformatics: A Practical Introduction
Chapman & Hall/CRC Press
2010
[10]
논문
An improved algorithm for matching biological sequences
https://linkinghub.e[...]
1982-12-15
[11]
논문
Multiple sequence alignment: Algorithms and applications
https://linkinghub.e[...]
1999-01-01
[12]
논문
On the complexity of multiple sequence alignment
[13]
논문
Settling the intractability of multiple alignment
[14]
논문
A tool for multiple sequence alignment
[15]
논문
CLUSTAL: a package for performing multiple sequence alignment on a microcomputer
[16]
논문
CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice
[17]
논문
Multiple sequence alignment with the Clustal series of programs
[18]
논문
T-Coffee: A novel method for fast and accurate multiple sequence alignment
[19]
논문
Comprehensive study on iterative algorithms of multiple sequence alignment
[20]
논문
Hidden Markov models for detecting remote protein homologies
[21]
논문
The relation between the divergence of sequence and structure in proteins
1986-04
[22]
논문
The protein structure prediction problem could be solved using the current PDB library
[23]
논문
Mapping the protein universe
[24]
논문
Multiple protein structure alignment
[25]
논문
CATH--a hierarchic classification of protein domain structures
[26]
논문
Protein structure alignment by incremental combinatorial extension (CE) of the optimal path
[27]
논문
Where Does the Alignment Score Distribution Shape Come from?
http://www.la-press.[...]
[28]
서적
Inferring Phylogenies
Sinauer Associates: Sunderland, MA
[29]
서적
Computer Methods for Macromolecular Sequence Analysis
[30]
논문
Sampling rare events: statistics of local sequence alignments
[31]
논문
Significance of gapped sequence alignments
[32]
논문
A probabilistic model of local sequence alignment that simplifies statistical significance estimation
[33]
논문
Fundamentals of massive automatic pairwise alignments of protein sequences: theoretical significance of Z-value statistics
[34]
논문
Pairwise Statistical Significance of Local Sequence Alignment Using Sequence-Specific and Position-Specific Substitution Matrices
[35]
논문
Pairwise statistical significance and empirical determination of effective gap opening penalties for protein local sequence alignment
http://inderscience.[...]
[36]
논문
Exact Calculation of Distributions on Integers, with Application to Sequence Alignment
[37]
서적
Bioinformatics
[38]
논문
Genome-wide identification of human RNA editing sites by parallel DNA capturing and sequencing
2009-05
[39]
논문
Whole genome assembly from 454 sequencing output via modified DNA graph concept
2009-06
[40]
논문
Single nucleotide polymorphism discovery in barley using autoSNPdb
2009-05
[41]
논문
Sequence Analysis and Optimal Matching Methods in Sociology, Review and Prospect
[42]
서적
Proceedings of the ACL-02 conference on Empirical methods in natural language processing - EMNLP '02
[43]
논문
Algorithms for Language Reconstruction
http://www.cs.ualber[...]
University of Toronto, Ontario
2007-01-21
[44]
논문
Incorporating sequential information into traditional classification models by using an element/position-sensitive SAM
http://econpapers.re[...]
[45]
웹사이트
ClustalW2 < Multiple Sequence Alignment < EMBL-EBI
http://www.ebi.ac.uk[...]
2017-06-12
[46]
웹사이트
T-coffee
https://web.archive.[...]
[47]
웹사이트
BLAST: Basic Local Alignment Search Tool
http://blast.ncbi.nl[...]
2017-06-12
[48]
웹사이트
UVA FASTA Server
http://fasta.bioch.v[...]
2017-06-12
[49]
논문
BAliBASE: a benchmark alignment database for the evaluation of multiple alignment programs
[50]
웹사이트
BAliBASE
https://web.archive.[...]
[51]
논문
A comprehensive comparison of multiple sequence alignment programs
[52]
웹사이트
Multiple sequence alignment: Strap
http://3d-alignment.[...]
2017-06-12
[53]
웹사이트
生命情報学(2)配列解析基礎
https://www.bic.kyot[...]
京都大学 化学研究所 バイオインフォマティクスセンター
2019
[54]
웹사이트
配列アライメント
https://dbarchive.bi[...]
DBCLS
[55]
논문
On the complexity of multiple sequence alignment
http://www.lieberton[...]
[56]
논문
Settling the intractability of multiple alignment
http://www.lieberton[...]
[57]
논문
A tool for multiple sequence alignment
http://www.pnas.org/[...]
[58]
논문
CLUSTAL: a package for performing multiple sequence alignment on a microcomputer
http://linkinghub.el[...]
[59]
논문
CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice
http://nar.oxfordjou[...]
[60]
논문
Multiple sequence alignment with the Clustal series of programs
http://nar.oxfordjou[...]
[61]
논문
T-Coffee: A novel method for fast and accurate multiple sequence alignment
http://linkinghub.el[...]
[62]
논문
Comprehensive study on iterative algorithms of multiple sequence alignment
http://bioinformatic[...]
[63]
논문
Hidden Markov models for detecting remote protein homologies
http://bioinformatic[...]
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.
문의하기 : help@durumis.com