BLAST (생명공학기술)
"오늘의AI위키"의 AI를 통해 더욱 풍부하고 폭넓은 지식 경험을 누리세요.
1. 개요
BLAST (Basic Local Alignment Search Tool)는 시퀀스 검색을 위한 생물정보학 프로그램으로, DNA 또는 단백질 시퀀스 간의 유사성을 빠르게 찾는 데 사용된다. 1990년 개발되었으며, 휴리스틱 알고리즘을 사용하여 대규모 게놈 데이터베이스에서 시퀀스 정렬을 효율적으로 수행한다. BLAST는 blastp, blastn, blastx, tblastn, tblastx 등 다양한 종류의 프로그램을 포함하며, 종 식별, 단백질 도메인 분석, 유전체 비교, 계통 발생 분석 등 다양한 생물학적 연구에 활용된다. BLAST의 대안으로는 FASTA, BLAT, HMMER 등이 있다.
더 읽어볼만한 페이지
- 생물정보학 알고리즘 - 서열 정렬
서열 정렬은 유전체 연구 및 생명과학에서 서열 간 진화적 관계를 밝히는 방법으로, 전역, 지역, 쌍별 정렬 등의 방법을 통해 계통수 분석, 유의성 및 신뢰도 평가 등에 활용되며, ClustalW2, BLAST, FASTA3x 등의 소프트웨어 도구로 수행된다. - 퍼블릭 도메인 소프트웨어 - 미스터리 하우스
미스터리 하우스는 1980년 출시된 어드벤처 게임으로, 그래픽을 도입하여 혁신을 시도했으며, 버려진 저택에서 단서를 찾아 살인자를 밝혀내는 내용을 담고 있다. - 퍼블릭 도메인 소프트웨어 - ANTLR
ANTLR은 EBNF로 표현된 문법을 입력받아 렉서, 파서, 트리 파서 등 다양한 언어 인식기 소스 코드를 생성하는 파서 생성기이며, C#, Java, Python 등 여러 언어를 지원하고 깃허브에 다양한 문법이 공개되어 있다. - 공식 웹사이트에 알 수 없는 변수를 사용한 문서 - 브루클린 미술관
브루클린 미술관은 1823년 브루클린 견습생 도서관으로 시작하여 현재 약 50만 점의 소장품을 보유한 뉴욕 브루클린 소재의 미술관으로, 다양한 분야의 예술 작품을 전시하며 특히 아프리카 미술과 여성주의 미술에 대한 기여가 크다. - 공식 웹사이트에 알 수 없는 변수를 사용한 문서 - 광주지방기상청
광주지방기상청은 광주광역시와 전라남도 지역의 기상 예보, 특보, 관측, 기후 정보 제공 등의 업무를 수행하는 기상청 소속 기관으로, 1949년 광주측후소로 설치되어 1992년 광주지방기상청으로 개편되었으며, 기획운영과, 예보과, 관측과, 기후서비스과와 전주기상지청, 목포기상대를 두고 있다.
| BLAST (생명공학기술) - [IT 관련 정보]에 관한 문서 | |
|---|---|
| 기본 정보 | |
| 종류 | 데이터베이스 검색 알고리즘 |
| 분야 | 생물정보학 |
| 개발자 | 스티븐 알tschul, 워렌 기쉬, 웹 밀러, 유진 마이어스, 데이비드 립먼 |
| 개발 | NCBI |
| 운영체제 | 유닉스, 리눅스, Mac, MS-Windows |
| 라이선스 | 퍼블릭 도메인 |
| 웹사이트 | BLAST 공식 웹사이트 |
| 기술 정보 | |
| 프로그래밍 언어 | C 및 C++ |
| 최신 버전 | 2.16.0+ |
| 최신 릴리스 날짜 | 2024년 6월 25일 |
2. 역사적 배경
BLAST는 1990년 미국 국립보건원 (NIH)의 Eugene Myers, Stephen Altschul, Warren Gish, David J. Lipman, Webb Miller에 의해 개발되었다.[5] 이들은 NIH에서 BLAST 프로그램을 설계하고 구현했다.[7]
BLAST는 1990년 ''Journal of Molecular Biology''에 게재된 논문을 통해 처음 소개되었으며, 현재까지 100,000회 이상 인용될 정도로 생명정보학 분야에서 널리 사용되는 알고리즘이다.[7]
BLAST는 단백질 및 DNA 서열 유사성 검색을 위해 이전에 개발된 프로그램인 FASTA의 기능을 확장했다.[5] Samuel Karlin과 Stephen Altschul이 개발한 새로운 확률적 모델을 추가하여,[5] "한 유기체의 알려진 DNA 시퀀스와 다른 유기체의 DNA 시퀀스 간의 유사성을 추정하는 방법"을 제안했다.[3] 이들의 연구는 "BLAST의 통계적 기초"로 묘사되었다.[6]
BLAST는 대부분의 경우 Smith-Waterman 구현보다 빠르지만, 최적의 정렬을 보장하지는 않는다. 대신 시퀀스에서 더 중요한 패턴만 검색하여 FASTA보다 시간 효율성이 높고 비교적 민감하다. 이러한 속도 강조는 현재 사용 가능한 거대한 게놈 데이터베이스에서 알고리즘을 실용적으로 만드는 데 필수적이었다.
BLAST의 원본 논문은 1990년 ''Journal of Molecular Biology''에 게재되었고,[7] 1990년대에 가장 많이 인용된 논문 중 하나였다.[10]
3. 알고리즘
BLAST 알고리즘은 크게 세 단계로 구성된다.
1. 질의 서열(query sequence)을 짧은 고정 길이(W)의 단편으로 분할하여 데이터베이스를 검색한다. 예를 들어 W=3이고, 질의 서열이 AGTTAC, 데이터베이스 서열이 ACTTAG라면, BLAST는 TTA라는 공통 부분을 인식한다.
2. BLAST는 질의 서열과 공통 부분을 공유하는 데이터베이스 서열에 대해 갭(gap)을 고려하지 않는 정렬을 수행하여, 정렬 점수가 높아지도록 양방향으로 확장한다.
3. 갭을 고려한 정렬을 수행하는 Smith-Waterman 알고리즘의 변형된 알고리즘을 통해, 통계적으로 유의미한 정렬 결과를 제공한다.
BLAST에서 파생된 알고리즘에는 BLAT와 BLASTZ 등이 있다. BLAT는 BLAST보다 빠르지만 정확도는 낮으며, BLASTZ는 대규모 게놈 데이터베이스 검색에 특화되어 있다.
BLAST 알고리즘을 실행하기 위해서는 질의 서열과 검색 대상 시퀀스 데이터베이스가 필요하다. BLAST는 데이터베이스에서 질의 서열과 유사한 서열을 찾는다. 일반적으로 질의 서열은 데이터베이스보다 훨씬 작다. 예를 들어, 질의 서열은 1,000개의 뉴클레오타이드이고 데이터베이스는 수십억 개의 뉴클레오타이드일 수 있다.
BLAST의 주요 아이디어는 통계적으로 유의미한 정렬에 종종 고득점 세그먼트 쌍(HSP)이 포함되어 있다는 것이다.
BLAST는 주로 시퀀스의 국소 정렬(로컬 정렬)을 수행하기 위해 사용된다. BLAST와 마찬가지로 짝을 이루는 정렬을 수행하기 위한 프로그램으로는 FASTA 등이 있다. 또한 다중 정렬을 수행하기 위한 프로그램으로는 Clustal (ClustalW, ClustalX) 등이 있다.
3. 1. 휴리스틱 알고리즘
BLAST는 휴리스틱 알고리즘을 사용하여 Smith-Waterman 구현보다 대부분의 경우 더 빠르지만, "질의 및 데이터베이스 시퀀스의 최적 정렬을 보장"할 수는 없다.[4] Smith-Waterman 알고리즘은 최상의 정렬을 찾을 수 있도록 보장된 최초의 시퀀스 정렬 알고리즘이었지만, 이러한 최적 알고리즘의 시간 및 공간 요구 사항은 BLAST의 요구 사항을 훨씬 초과한다.
BLAST는 시퀀스에서 더 중요한 패턴만 검색하여 FASTA보다 시간 효율성이 더 높지만 비교적 민감하다.
휴리스틱 방법을 사용하여 BLAST는 두 시퀀스 간의 짧은 일치를 찾아 유사한 시퀀스를 찾는다. 유사한 시퀀스를 찾는 이 과정을 시딩이라고 한다. BLAST가 로컬 정렬을 시작하는 것은 이 첫 번째 일치 이후이다. 시퀀스의 유사성을 찾으려고 할 때, 단어라고 알려진 공통 문자 집합이 매우 중요하며, 정상적인 조건에서 단어 크기는 3자가 된다. BLAST의 휴리스틱 알고리즘은 관심 시퀀스와 데이터베이스의 히트 시퀀스 또는 시퀀스 간의 모든 공통 세 글자 단어를 찾는다. 이 결과는 정렬을 구축하는 데 사용된다. 관심 시퀀스에 대한 단어를 만든 후, 나머지 단어들도 조립된다. 이러한 단어는 점수 행렬을 사용하여 비교할 때 최소 임계값 ''T'' 이상의 점수를 가져야 한다.
BLAST 검색에 일반적으로 사용되는 점수 행렬 중 하나는 BLOSUM62이다.[11] 최적의 점수 행렬은 시퀀스 유사성에 따라 다르지만, 일단 단어와 인접 단어가 조립되고 컴파일되면, 일치를 찾기 위해 데이터베이스의 시퀀스와 비교된다. 임계값 점수 ''T''는 특정 단어가 정렬에 포함될지 여부를 결정한다. 시딩이 수행되면, 3개의 잔기로만 이루어진 정렬은 BLAST에서 사용되는 알고리즘에 의해 양쪽 방향으로 확장된다. 각 확장은 정렬의 점수에 영향을 미쳐 점수를 증가시키거나 감소시킨다. 이 점수가 미리 결정된 ''T''보다 높으면, 정렬은 BLAST에서 제공하는 결과에 포함된다. 그러나 이 점수가 이 미리 결정된 ''T''보다 낮으면, 정렬은 확장을 중단하여, 불량한 정렬 영역이 BLAST 결과에 포함되지 않도록 한다. ''T'' 점수를 증가시키면 검색에 사용할 수 있는 공간의 양이 제한되고 인접 단어의 수가 감소하는 동시에 BLAST 프로세스의 속도가 빨라진다.
BLAST의 주요 아이디어는 통계적으로 유의미한 정렬에 종종 고득점 세그먼트 쌍(HSP)이 포함되어 있다는 것이다. BLAST는 쿼리 시퀀스와 데이터베이스의 기존 시퀀스 간의 고득점 시퀀스 정렬을 Smith-Waterman 알고리즘을 근사하는 휴리스틱 접근 방식을 사용하여 검색한다. 그러나 Smith-Waterman의 완전 탐색 접근 방식은 GenBank와 같은 대규모 유전체 데이터베이스를 검색하기에는 너무 느리다. 따라서 BLAST 알고리즘은 Smith-Waterman 알고리즘보다 정확도는 떨어지지만 50배 이상 빠른 휴리스틱 알고리즘 접근 방식을 사용한다.[12] BLAST의 속도와 상대적으로 좋은 정확도는 BLAST 프로그램의 주요 기술 혁신 중 하나이다.
3. 2. Smith-Waterman 알고리즘과의 비교
BLAST와 Smith-Waterman 알고리즘은 모두 질의 서열과 데이터베이스 서열을 비교하여 상동 서열을 찾지만, 몇 가지 차이점이 있다.
BLAST는 휴리스틱 알고리즘을 기반으로 하므로, BLAST를 통해 얻은 결과는 데이터베이스 내의 모든 가능한 일치를 포함하지는 않는다. 즉, BLAST는 찾기 어려운 일치를 놓칠 수 있다.
모든 가능한 일치를 찾기 위한 대안은 Smith-Waterman 알고리즘을 사용하는 것이다. 이 방법은 정확도와 속도라는 두 가지 측면에서 BLAST 방법과 다르다. Smith-Waterman 알고리즘은 모든 정보를 배제하지 않기 때문에 BLAST가 찾을 수 없는 일치를 찾아내어 더 나은 정확도를 제공한다. 따라서 원격 상동성(remote homology) 검색에 필요하다. 그러나 BLAST와 비교했을 때, 시간이 더 오래 걸리고 많은 양의 컴퓨팅 성능과 메모리가 필요하다. 하지만, FPGA 칩과 SIMD 기술을 활용하여 Smith-Waterman 검색 프로세스의 속도를 획기적으로 높이기 위한 발전이 이루어졌다.
BLAST에서 더 완전한 결과를 얻으려면 설정을 기본 설정에서 변경할 수 있다. 그러나 주어진 서열에 대한 최적의 설정은 다를 수 있다. 변경할 수 있는 설정에는 E-value, 갭 비용, 필터, 단어 크기 및 치환 행렬이 있다.[28]
4. BLAST의 종류
BLAST는 생물정보학에서 DNA의 염기 서열 또는 단백질의 아미노산 서열 시퀀스에 대해 짝을 이루는 시퀀스 정렬을 수행하는 프로그램이다. blastall 실행 파일에는 blastp, blastn, blastx, tblastn, tblastx 등 여러 프로그램이 포함되어 있으며, 명령줄 환경에서 -p 옵션으로 실행할 프로그램을 지정한다(예: blastall -p blastn).[14][15][16][17] BLAST 프로그램을 실행할 때는 FASTA 형식으로 쿼리 시퀀스를 전달한다.
주요 BLAST 프로그램은 다음과 같다:
- blastp: 아미노산 서열 (단백질의 시퀀스)을 쿼리로 하여, 지정한 아미노산 서열 데이터베이스에서 유사한 서열을 검색한다.
- blastn: 염기 서열 (뉴클레오티드의 시퀀스)을 쿼리 시퀀스로 하여, 지정한 염기 서열 데이터베이스에서 유사한 서열을 검색한다.
- blastx: 염기 서열의 쿼리 시퀀스를 6개의 프레임으로 번역하여, 아미노산 서열 데이터베이스에서 유사한 시퀀스를 검색한다.
- tblastn: 아미노산 서열을 쿼리로 하여, 염기 서열 데이터베이스를 6개의 프레임으로 번역하면서 유사한 시퀀스를 검색한다.
- tblastx: 염기 서열의 쿼리 시퀀스를 6개의 프레임으로 번역하고, 아미노산 서열로 번역된 염기 서열 데이터베이스에서 유사한 시퀀스를 검색한다. 처리 속도가 가장 느리며, 염기 서열 간의 매우 먼 관련성을 발견하는 데 사용된다.
이 외에도 PSI-BLAST, megablast, BLAT, BLASTZ와 같은 파생 알고리즘 및 프로그램이 존재한다. PSI-BLAST는 단백질의 먼 계통적 관련성을 발견하는 데 사용되고, megablast는 대량의 쿼리 시퀀스로 고속 검색을 수행하며, BLAT는 BLAST보다 빠르지만 정확도는 낮고, BLASTZ는 대규모 게놈 데이터베이스 검색에 사용된다.
4. 1. blastn
BLASTn은 뉴클레오타이드 서열을 사용하여 데이터베이스 또는 다른 뉴클레오타이드 서열을 검색한다. 이는 유기체 간의 진화적 관계를 식별하는 데 유용하다.[14]4. 2. blastp
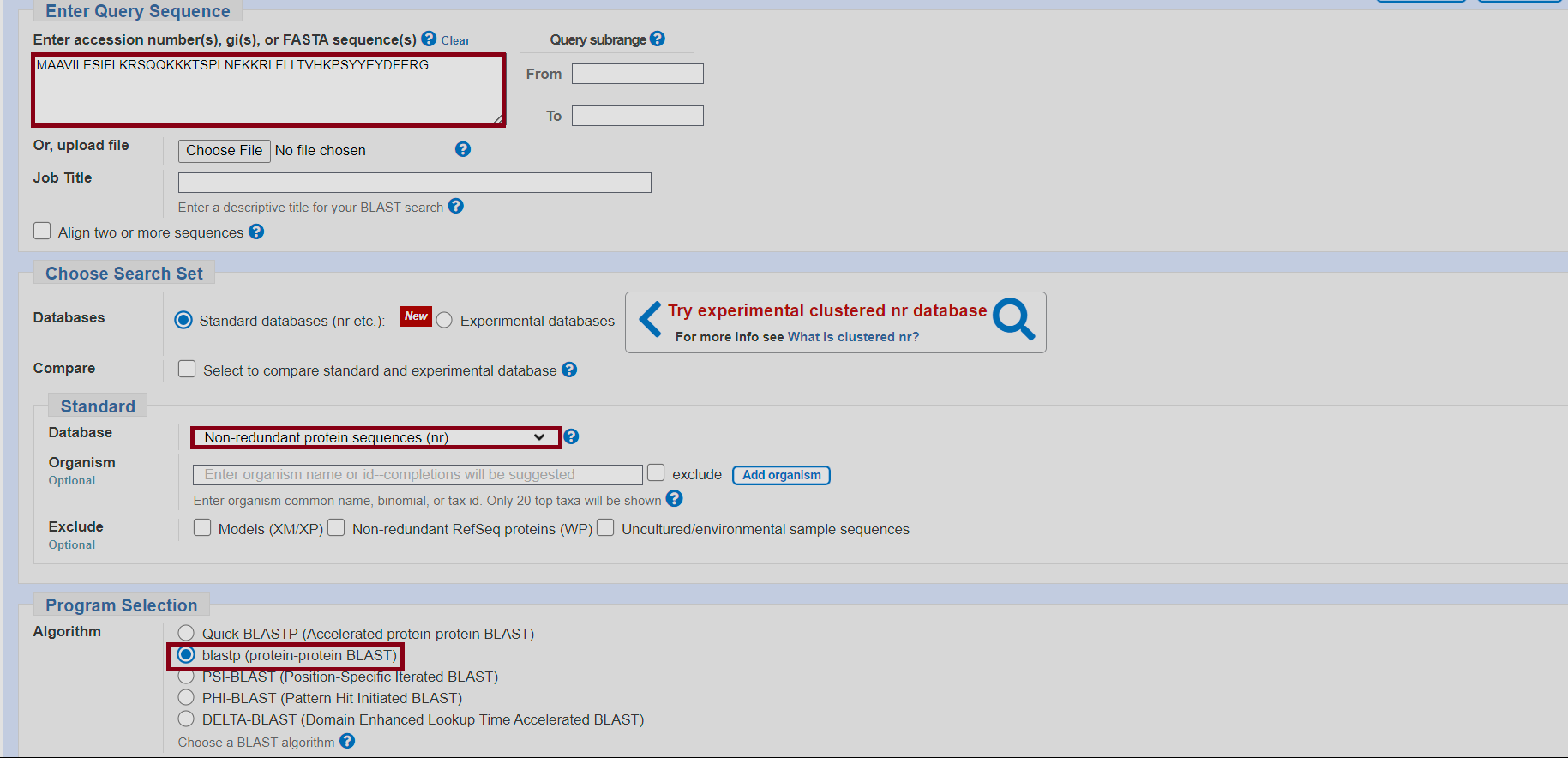
BLASTp (단백질 BLAST)는 단백질 서열을 비교하는 데 사용된다. 단일 단백질 서열이나, 비교하려는 하나 이상의 단백질 서열을 데이터베이스와 비교할 수 있다. 이는 기존 단백질 데이터베이스에서 유사한 서열을 찾아 단백질을 식별하려는 경우에 유용하다.[17]
4. 3. blastx
BLASTx는 뉴클레오타이드 쿼리 서열을 여섯 개의 가능한 단백질 서열로 번역하여 알려진 단백질 서열 데이터베이스와 비교한다.[16] 이 도구는 DNA 서열의 판독 프레임이 불확실하거나, 단백질 코딩에 오류를 일으킬 수 있는 오류가 포함된 경우에 유용하다.[16] BLASTx는 모든 프레임에서 히트에 대한 결합된 통계를 제공하여 새로운 DNA 서열의 초기 분석에 도움을 주며, 유전자의 코딩 영역(coding region)을 예측하는데 사용된다.[16]4. 4. tblastn
tblastn은 단백질로 아직 번역되지 않은 서열에서 단백질을 검색하는 데 사용된다. 단백질 서열을 가져와 DNA 서열의 모든 가능한 번역과 비교한다. 이는 EST(짧은 단일 가닥 cDNA 서열) 및 HTG(초안 게놈 서열)와 같이 완전히 주석 처리되지 않은 DNA 서열에서 유사한 단백질 코딩 영역을 찾을 때 유용하다. 이러한 서열은 알려진 단백질 번역이 없으므로 tblastn을 사용하여 검색할 수 있다.[15]4. 5. tblastx
주어진 원본 소스에는 'tblastx'에 대한 정보가 없으므로, 해당 섹션 내용을 작성할 수 없습니다. 이전 답변과 동일하게 빈 문자열을 출력합니다.5. 병렬 BLAST
병렬 BLAST는 대규모 서열 데이터베이스 검색 시간을 단축하기 위해 개발되었다. 병렬 BLAST는 여러 프로세서 또는 컴퓨터를 사용하여 검색을 동시에 수행한다. 데이터베이스는 동일한 크기로 분할되어 각 노드에 로컬로 저장된다. 각 쿼리는 모든 노드에서 병렬로 실행되며, 모든 노드에서 생성된 BLAST 출력 파일이 병합되어 최종 출력을 생성한다. MPIblast, ScalaBLAST, DCBLAST 등이 병렬 BLAST의 구체적인 구현 예시이다.[18]
MPIblast는 데이터베이스 분할 기술을 활용하여 계산 프로세스를 병렬화한다.[19] 이를 통해 클러스터의 여러 노드에서 BLAST 검색을 수행할 때 상당한 성능 향상을 얻을 수 있으며, 일부 시나리오에서는 초선형 속도 향상도 가능하다. 이는 MPIblast를 생물 정보학에서 일반적으로 사용되는 광범위한 유전체 데이터 세트에 적합하게 만든다.
BLAST는 일반적으로 데이터베이스 크기가 n일 때 O(n) 속도로 실행된다.[20] 검색을 완료하는 시간은 데이터베이스 크기가 증가함에 따라 선형적으로 증가한다. MPIblast는 병렬 처리를 활용하여 검색 속도를 높인다. 모든 병렬 계산에 대한 이상적인 속도는 복잡도 O(n/p)이며, 여기서 n은 데이터베이스 크기이고 p는 프로세서 수이다. 이는 작업이 p개의 프로세서에 균등하게 분산됨을 나타낸다. MPIblast에서 때때로 발생할 수 있는 초선형 속도 향상은 O(n/p)보다 나은 복잡성을 가질 수 있다. 이는 캐시 메모리를 사용하여 실행 시간을 줄일 수 있기 때문에 발생한다.[21]
병렬 처리나 분산 처리 환경에 대응하는 BLAST 알고리즘이 몇 가지 고안되었다. 이러한 알고리즘은 일반적으로 Parallel BLAST라고 불리는 경우가 있다. Parallel BLAST는 Pthreads나 MPI의 API를 사용하여 구현된다. 리눅스, Windows, 솔라리스, AIX 등 많은 컴퓨터 플랫폼에 이식되어 사용 가능하다.
BLAST 알고리즘을 병렬 처리나 분산 처리에 대응시키는 방법은 다음과 같다.
- 분산 쿼리
- 해시 테이블 분할
- 계산 처리 병렬화
- 데이터베이스 분할
6. BLAST의 활용
BLAST는 종 식별, 도메인 위치, 계통 발생 확립, DNA 매핑, 유전자 비교 등 여러 가지 목적으로 사용될 수 있다.
BLAST는 생물정보학에서 DNA의 염기 서열 또는 단백질의 아미노산 서열에 대해 짝을 이루는 시퀀스 정렬을 수행하며, 주로 서열의 국소 정렬(로컬 정렬)을 위해 사용된다. BLAST와 마찬가지로 짝을 이루는 정렬을 수행하는 프로그램에는 FASTA 등이 있다. 또한 다중 정렬을 수행하는 프로그램에는 Clustal (ClustalW, ClustalX) 등이 있다.
BLAST는 생물정보학에서 가장 널리 사용되는 프로그램 중 하나인데, 그 이유는 BLAST가 처리 속도를 매우 중요하게 생각하기 때문이다. BLAST의 알고리즘은 정확도보다 속도를 중시하며, 이러한 속도 중시는 오늘날 방대한 데이터가 축적되어 있는 게놈의 시퀀스 데이터베이스에 대해 검색을 수행할 때 BLAST 알고리즘을 실용적인 것으로 만든다. BLAST가 개발된 후 BLAST보다 빠른 알고리즘이 몇 가지 개발되었다. BLAST는 대략적인 시퀀스 매칭을 필요로 하는 다른 알고리즘의 일부로도 자주 사용된다.
BLAST를 사용하면, 예를 들어 생쥐에서 미지의 유전자를 발견했을 때, 사람이 해당 시퀀스와 유사한 유전자를 가지고 있는지 조사할 수 있다. 생쥐의 미지 시퀀스로 BLAST를 사용하여 사람 게놈의 시퀀스 데이터베이스에 대해 검색을 수행하면, BLAST는 유사성이 높은 시퀀스 군을 찾아낸다.
또한 BLAST는 다음과 같은 질문에 답하는 데 사용될 수 있다.
- 손에 있는 단백질의 아미노산 서열은 어떤 박테리아와 계통적으로 관련이 있는가?
- 지금 시퀀싱하여 얻은 DNA는 어떤 종에서 유래하는가?
- 자신이 결정한 구조(또는 구조 모티프)를 가진 단백질을 기록한 다른 유전자가 있는가?
6. 1. 종 식별
BLAST를 사용하면 종을 정확하게 식별하거나 동족 종을 찾을 수 있다. 이는 예를 들어 알려지지 않은 종의 DNA 염기서열을 다룰 때 유용하다.[30]6. 2. 유전자 발견 및 주석
BLAST는 두 개의 관련된 종에서 공통 유전자를 찾아 한 유기체에서 다른 유기체로 유전자에 주석을 다는 데 사용될 수 있다.6. 3. 계통 발생 분석
BLAST를 통해 얻은 결과로 BLAST 웹 페이지에서 계통 발생 트리를 만들 수 있다. 그러나 BLAST만을 기반으로 한 계통 발생은 다른 목적으로 만들어진 계산적 계통 발생학 방법에 비해 신뢰성이 떨어지므로, "첫 번째 통과" 계통 발생 분석에만 의존해야 한다.[30]BLAST는 유전 염기서열을 사용하여 알려진 분류학적 데이터에 대해 여러 분류군을 비교할 수 있다. 이를 통해 다양한 종 간의 진화적 관계를 보여줄 수 있다(그림 6). 이는 고아 유전자를 식별하는 데 유용한데, 유전자가 조상 계통 밖의 유기체에서 나타나는 경우 고아 유전자로 분류되지 않기 때문이다.
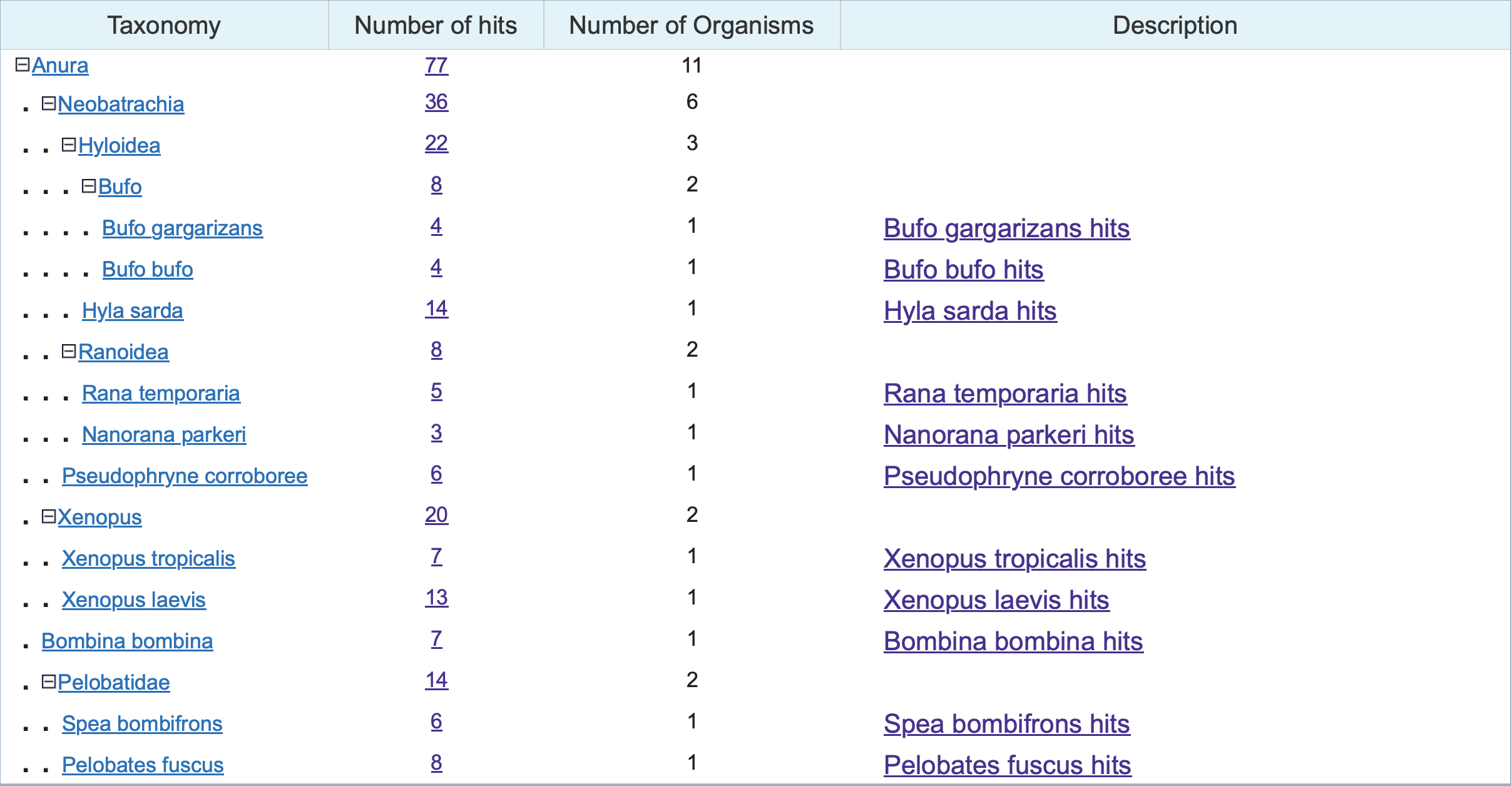
이 방법이 도움이 되지만, 상동체를 찾기 위한 더 정확한 옵션으로는 쌍을 이루는 염기서열 정렬과 다중 염기서열 정렬이 있다.
6. 4. 단백질 도메인 분석
BLAST는 단백질 서열을 다룰 때, 관심 있는 서열 내에서 알려진 단백질 도메인을 찾는 데 사용될 수 있다.[30]6. 5. 유전체 비교
BLAST는 서로 다른 종의 유전자를 비교하여 공통 유전자를 찾고, 한 유기체의 주석을 다른 유기체로 매핑하는 데 사용될 수 있다. 또한, 유전 염기서열을 사용하여 알려진 분류학적 데이터에 대해 여러 분류군을 비교함으로써 다양한 종 간의 진화적 관계를 보여줄 수 있다. BLASTP영어 검색 결과, Bufo japonicus에서 발견된 유전자가 개구리 계통의 다른 많은 종에서도 발견됨을 확인할 수 있다.이러한 방법 외에도 상동체를 찾기 위한 더 정확한 옵션으로는 쌍을 이루는 염기서열 정렬과 다중 염기서열 정렬이 있다.
7. BLAST 대안
BLAST 외에도 다양한 서열 정렬 도구가 존재한다.
PatternHunter는 BLAT과 유사한 소프트웨어이다.
2000년대 후반 시퀀싱 기술 발달로 매우 유사한 뉴클레오티드 일치 항목 검색이 중요해졌다. 이를 위해 개발된 정렬 프로그램들은 대상 데이터베이스(주로 게놈)의 BWT 인덱싱을 사용한다. 입력 서열을 빠르게 매핑하고, 출력은 BAM 파일 형식이다. 예시로는 BWA, SOAP, Bowtie가 있다.
PLAST는 두 서열 뱅크 비교를 위한 BLAST의 대안이다. PLAST[23] 및 ORIS[24] 알고리즘에 의존하는 고성능 범용 뱅크 대 뱅크 서열 유사성 검색 도구로, BLAST와 결과는 유사하지만 훨씬 빠르고 적은 메모리로 대량 서열 비교가 가능하다.
MMseqs는 오픈 소스 소프트웨어로, BLAST/PSI-BLAST의 대안이다. 속도-민감도 균형에서 기존 도구를 개선하여 PSI-BLAST보다 400배 이상 빠른 속도로 더 나은 민감도를 달성한다.[26]
광학 컴퓨팅 접근법은 현재 전기적 구현의 대안으로 제시되었으며, OptCAM은 그러한 예시로 BLAST보다 빠르다.[27]
7. 1. FASTA
BLAST의 전신인 FASTA는 단백질 및 DNA 유사성 검색에 사용될 수 있다. FASTA는 단백질과 단백질, DNA와 DNA 및 단백질 데이터베이스를 비교하기 위한 유사한 일련의 프로그램을 제공하며, 정렬되지 않은 짧은 펩타이드와 DNA 서열을 처리하기 위한 추가 프로그램도 포함한다. 또한, FASTA 패키지는 엄격한 스미스-워터만 알고리즘의 벡터화된 구현인 SSEARCH를 제공한다. FASTA는 BLAST보다 느리지만 훨씬 더 광범위한 점수 행렬을 제공하여 특정 진화적 거리에 맞춰 검색을 용이하게 한다.[22]7. 2. BLAT
BLAT(Blast Like Alignment Tool, 블라스트 유사 정렬 도구)는 BLAST보다 빠르지만 감도가 낮은 서열 정렬 도구이다.[22] BLAST가 선형 검색을 수행하는 반면, BLAT는 데이터베이스의 k-mer 인덱싱에 의존하므로 종종 더 빠르게 시드를 찾을 수 있다. BLAT은 뉴클레오티드 서열로 게놈 데이터베이스 검색을 수행한다.7. 3. HMMER
은닉 마르코프 모델을 사용하여 알려진 도메인(Pfam에서)을 검색하는 데에는 HMMER와 같은 인기 있는 대안이 있다.[22]7. 4. DIAMOND
DIAMOND[25]는 메타게노믹스 응용 분야에서 BLASTX보다 최대 20,000배 빠르게 실행되면서도 높은 수준의 민감도를 유지하는 도구이다. 이는 수십억 개의 짧은 DNA 리드를 수천만 개의 단백질 참조와 비교해야 하는 과제에 유용하다.8. BLAST 출력 가시화
사용자가 BLAST 결과를 해석하는 데 도움을 주기 위해 다양한 소프트웨어가 제공된다. 설치 및 사용, 분석 기능 및 기술에 따라 사용 가능한 도구는 다음과 같다.[29]
| 종류 | 도구 |
|---|---|
| NCBI BLAST 서비스 | NCBI BLAST 서비스 |
| 일반적인 BLAST 결과 해석기, GUI 기반 | JAMBLAST, Blast Viewer, BLASTGrabber |
| 통합 BLAST 환경 | PLAN, BlastStation-Free, [https://SequenceServer.com SequenceServer] |
| BLAST 결과 파서 | MuSeqBox, Zerg, BioParser, BLAST-Explorer, [https://SequenceServer.com SequenceServer] |
| 특수 BLAST 관련 도구 | [https://www.wsi.uni-tuebingen.de/lehrstuehle/algorithms-in-bioinformatics/software/megan6/ MEGAN], BLAST2GENE, BOV, [http://tools.bat.infspire.org/circoletto/ Circoletto] |
BLAST 결과의 시각화 예시는 그림 4와 5에 나와 있다.
참조
[1]
웹사이트
BLAST Developer Information
https://blast.ncbi.n[...]
[2]
서적
BLAST Release Notes
https://www.ncbi.nlm[...]
National Center for Biotechnology Information (US)
2024-06-24
[3]
뉴스
Samuel Karlin, Versatile Mathematician, Dies at 83
https://www.nytimes.[...]
2008-02-21
[4]
웹사이트
BLAST Sequences Aid in Genomics and Proteomics
http://www.b-eye-net[...]
Business Intelligence Network
[5]
웹사이트
BLAST topics
https://www.ncbi.nlm[...]
[6]
웹사이트
Sam Karlin, mathematician who improved DNA analysis, dead at 83
https://news.stanfor[...]
2019-07-16
[7]
학술지
Basic local alignment search tool
http://www.blastalgo[...]
[8]
학술지
ScalaBLAST: A Scalable Implementation of BLAST for High-Performance Data-Intensive Bioinformatics Analysis
https://zenodo.org/r[...]
[9]
학술지
ScalaBLAST 2.0: Rapid and robust BLAST calculations on multiprocessor systems
[10]
웹사이트
Sense from Sequences: Stephen F. Altschul on Bettering BLAST
http://www.sciencewa[...]
ScienceWatch
2000-07
[11]
학술지
Amino Acid Substitution Matrices from Protein Blocks
[12]
서적
Bioinformatics: Sequence and Genome Analysis
http://www.bioinform[...]
Cold Spring Harbor Press
[13]
Youtube
Adapted from Biological Sequence Analysis I, Current Topics in Genome Analysis
https://www.youtube.[...]
[14]
웹사이트
Library Guides: NCBI Bioinformatics Resources: An Introduction: BLAST: Compare & identify sequences
https://guides.lib.b[...]
[15]
웹사이트
Library Guides: NCBI Bioinformatics Resources: An Introduction: BLAST: Compare & identify sequences
https://guides.lib.b[...]
[16]
웹사이트
Library Guides: NCBI Bioinformatics Resources: An Introduction: BLAST: Compare & identify sequences
https://guides.lib.b[...]
[17]
웹사이트
Library Guides: NCBI Bioinformatics Resources: An Introduction: BLAST: Compare & identify sequences
https://guides.lib.b[...]
[18]
학술지
Divide and Conquer (DC) BLAST: fast and easy BLAST execution within HPC environments
2017
[19]
웹사이트
The design, implementation, and evaluation of mpiBLAST
https://pages.cs.wis[...]
2023-04-17
[20]
웹사이트
The Blast Algorithm (Basic Alignment Search Tool
https://bio.libretex[...]
2023-04-17
[21]
웹사이트
The design, implementation, and evaluation of mpiBLAST
https://pages.cs.wis[...]
2023-04-17
[22]
학술지
BLAT—The BLAST-Like Alignment Tool
2002-04-01
[23]
학술지
PLAST: parallel local alignment search tool for database comparison
[24]
서적
2008 IEEE International Symposium on Parallel and Distributed Processing
https://hal.archives[...]
[25]
학술지
Fast and sensitive protein alignment using DIAMOND
2015
[26]
학술지
MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets
2017-10-16
[27]
학술지
OptCAM: An ultra-fast all-optical architecture for DNA variant discovery
[28]
웹사이트
Bioinformatics Explained: BLAST versus Smith-Waterman
http://www.montefior[...]
2007-07-04
[29]
학술지
BLAST output visualization in the new sequencing era
2014
[30]
웹사이트
NCBI Magic-BLAST
https://ncbi.github.[...]
2019-05-16
[31]
웹인용
BLAST Developer Information
https://blast.ncbi.n[...]
[32]
서적
BLAST Release Notes
https://www.ncbi.nlm[...]
National Center for Biotechnology Information (US)
2023-10-24
[33]
뉴스
Samuel Karlin, Versatile Mathematician, Dies at 83
https://www.nytimes.[...]
2008-02-21
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.
문의하기 : help@durumis.com