뉴클레오사이드와 뉴클레오타이드 역전사효소 억제제의 발견과 발전
"오늘의AI위키"의 AI를 통해 더욱 풍부하고 폭넓은 지식 경험을 누리세요.
1. 개요
뉴클레오사이드와 뉴클레오타이드 역전사효소 억제제는 후천면역결핍증후군(AIDS)을 유발하는 인간면역결핍 바이러스(HIV) 감염 치료에 사용되는 약물이다. 이 계열의 약물은 HIV-1의 역전사 효소를 억제하여 바이러스의 복제를 막는 기전을 가지며, 지도부딘을 시작으로 디다노신, 잘시타빈, 라미부딘, 테노포비르 등 다양한 약물이 개발되었다. 역전사 효소는 바이러스 RNA를 DNA로 변환하는 역할을 하며, 뉴클레오사이드 및 뉴클레오타이드 역전사 효소 억제제는 이 과정에 필요한 물질의 유사체로 작용하여 DNA 사슬의 합성을 방해한다. HIV의 약물 저항성 문제를 해결하기 위해 HAART 요법과 같은 약물 조합이 사용되며, 아프리시타빈, 엘부시타빈, 암독소비르, 라시비르 등 새로운 약물들이 개발 중에 있다.
더 읽어볼만한 페이지
- 항레트로바이러스제 - 마라비록
마라비록은 CCR5-트로픽 HIV-1 감염 치료에 사용되는 진입 억제제 계열의 항레트로바이러스 약물로, CCR5 수용체 결합을 통해 HIV의 세포 진입을 막고 CYP3A에 의해 대사되며, 심각한 간 질환 등의 부작용이 있을 수 있고, 백혈병 환자의 이식편대숙주병 감소 효과에 대한 연구가 진행 중이며, 화이자에 의해 개발되어 2007년 미국 FDA와 유럽 연합에서 승인받았다. - 항레트로바이러스제 - 잘시타빈
잘시타빈은 제롬 호르비츠가 합성한 피리미딘 유사체 항레트로바이러스제로, HIV 치료제로 FDA 승인을 받았으나, 뉴클레오시드 유사체 역전사효소 저해제 병용 요법의 등장으로 2006년 판매 및 유통이 중단되었다.
| 뉴클레오사이드와 뉴클레오타이드 역전사효소 억제제의 발견과 발전 | |
|---|---|
| 약물 정보 | |
| ATC 코드 | J05A |
| 법적 지위 | 처방전 필요 |
| 역사 | |
| 발견 접근 방식 | 구조 기반 약물 설계 합리적 약물 설계 약물 스크리닝 |
| 주요 개발 단계 | 1964년: 티미딘 유사체의 항바이러스 활성 발견 1985년: 지도부딘이 HIV에 효과적인 것으로 입증됨 1987년: 지도부딘이 FDA 승인을 받음 1996년: HAART 요법 도입 |
| 작용 기전 | |
| 작용 방식 | 역전사효소 억제 DNA 사슬 종결 |
| 약물 종류 | |
| 주요 종류 | NRTI NtRTI |
| 예시 | |
| NRTI | 지도부딘 라미부딘 엠트리시타빈 아바카비르 디다노신 스타부딘 |
| NtRTI | 테노포비르 |
2. 역사
1964년, 지도부딘(AZT)은 미시간 암 재단(Michigan Cancer Foundation)의 호르비츠(Horwitz)에 의해 처음 합성되었다. 이는 티미딘의 데옥시리보스 고리 3' 위치 수산기(-OH)를 아지도기(-N3)로 대체한 화합물이었다.[13] 지도부딘은 원래 잠재적인 항암제로 개발되었으나, 효과가 없는 것으로 밝혀졌다.[15] 이후 1974년, 지도부딘이 레트로바이러스에 대한 활성을 보인다는 연구 결과가 보고되었다.[13]
HIV-1 역전사효소(RT)는 HIV-1 바이러스 복제에 필수적인 효소이다. 이 효소는 비대칭적인 헤테로다이머(heterodimer) 구조를 가지며, p66과 p51이라는 두 개의 서로 다른 단백질 소단위체(subunit)로 구성된다.[8][37] 더 큰 소단위체인 p66은 효소의 주요 활성을 나타내며[8], 더 작은 소단위체인 p51은 구조적 안정성에 기여하는 것으로 여겨진다.[8][37]
1981년 여름, 최초로 후천면역결핍증후군(AIDS) 사례가 보고되었고,[30] 2년 뒤인 1983년에는 AIDS의 원인 바이러스인 인간면역결핍 바이러스(HIV)가 발견되었다.[31][32] HIV의 정체가 밝혀진 후, 효과적인 항레트로바이러스(antiretroviral) 의약품 개발과 HIV에 대한 방대한 연구가 지속되었다.[32][33] 특히 HIV-1 바이러스의 역전사 효소는 항-HIV 의약품 개발의 주요 표적이 되었다.[34]
1980년대 중반 서구 사회에 AIDS가 유행하면서, 이전에 레트로바이러스 활성이 보고되었던 지도부딘이 항바이러스제로서 다시 주목받게 되었다.[13] 지도부딘은 역전사 과정에서 DNA 사슬이 신장될 때, 다음 뉴클레오타이드가 결합하는 데 필요한 3' 수산기가 없기 때문에 DNA 합성을 중단시키는 사슬 종결자(chain terminator)로 작용한다. 즉, 티미딘 대신 DNA 사슬에 삽입되어 HIV 복제를 강력하게 억제한다.[14]
체외(in vitro) 실험에서 항-HIV 효과를 보인 최초의 뉴클레오사이드 역전사효소 억제제(NRTI)인 지도부딘은 1987년 미국 식품의약국(FDA)의 승인을 받았다.[35] 지도부딘 승인 이후, 여러 NRTI와 하나의 뉴클레오타이드 역전사효소 억제제(NtRTI)가 FDA 승인을 받았다. 승인된 NRTI에는 지도부딘 외에 디다노신(didanosine), 잘시타빈(zalcitabine), 스타부딘(stavudine), 라미부딘(lamivudine), 아바카비르(abacavir), 엠트리시타빈(emtricitabine)이 있으며, 유일하게 승인된 NtRTI는 테노포비르(tenofovir)이다.[33][35]
현재 HIV 감염 치료에 사용되는 항레트로바이러스 약물은 크게 여섯 가지 계열로 분류된다: 뉴클레오사이드 및 뉴클레오타이드 역전사효소 억제제(NRTI/NtRTI), 비뉴클레오사이드 역전사 효소 억제제(NNRTI), 단백질분해효소 억제제(PI), 진입 억제제, 공동-수용체 억제제(co-receptor inhibitor), 그리고 통합효소 억제제(integrase inhibitor)이다.[33]
한편, 지도부딘은 비교적 독성이 있는 약물로 알려져 있는데, 이는 세포 내 효소에 의해 삼인산염 형태로 활성화될 때 감염되지 않은 정상 세포에도 영향을 미칠 수 있기 때문이다.
3. HIV-1 역전사 효소
3. 1. 기능
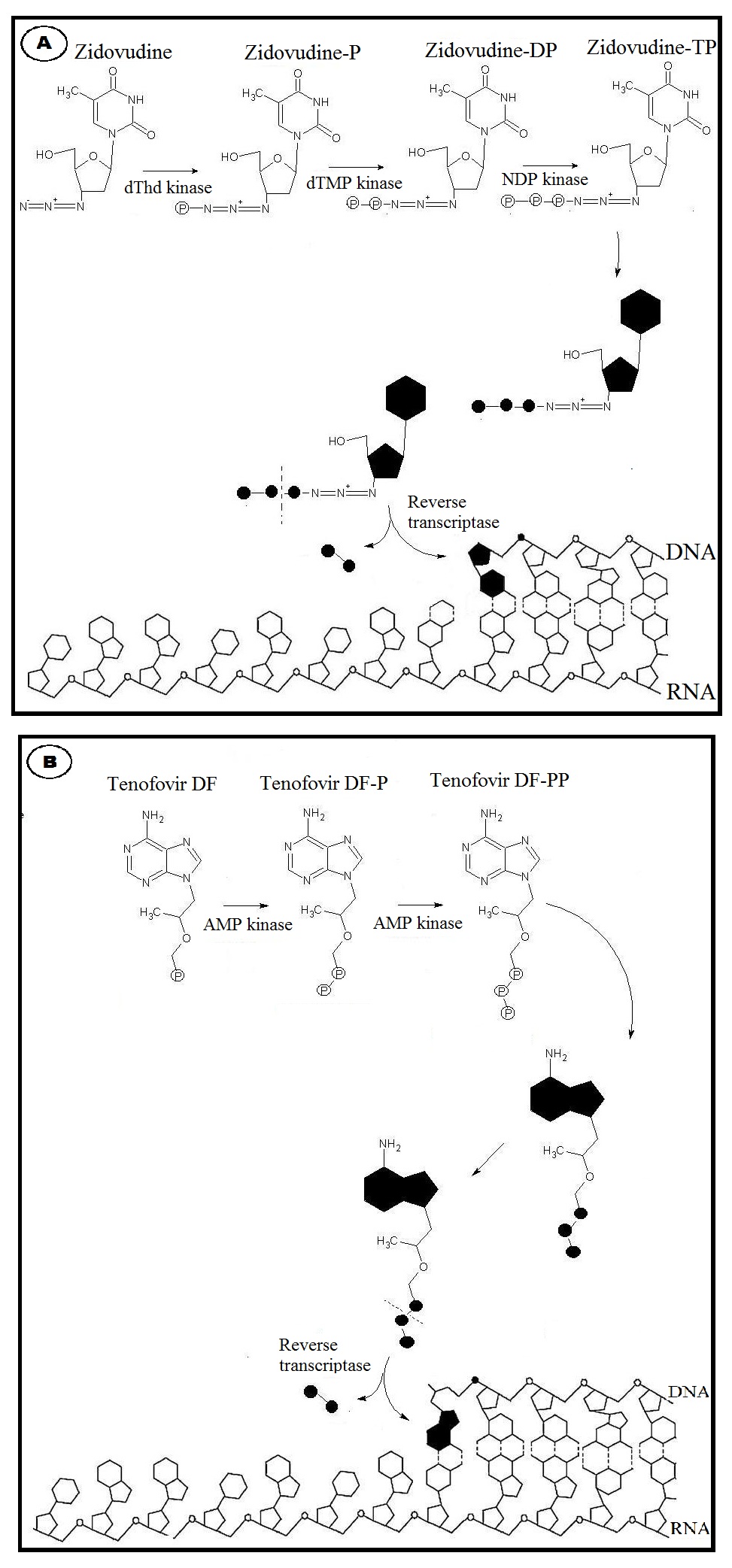
대부분의 표준 HIV 약물 치료법은 역전사 효소(Reverse Transcriptase, RT)를 억제하는 데 초점을 맞춘다. 역전사 효소는 HIV-1을 비롯한 다른 레트로바이러스가 자신의 생활사를 완성하기 위해 필수적인 효소이다.[34][5]
역전사 효소는 두 가지 주요 기능을 수행한다.
# '''중합효소 활성''': 바이러스의 유전 물질 복제를 담당한다. 바이러스의 단일 가닥 RNA를 숙주 게놈에 삽입될 수 있는 이중 가닥 DNA로 전환시킨다. 이렇게 생성된 DNA는 숙주 세포의 핵으로 이동하여 레트로바이러스의 인테그라제(integrase) 효소에 의해 숙주 게놈에 통합된다.[34][7]
# '''리보핵산 분해효소 H 활성''': RNA가 DNA와 헤테로듀플렉스(heteroduplex)를 형성했을 때 RNA 가닥을 분해한다.[36][37][8]
뉴클레오사이드 및 뉴클레오타이드 역전사효소 억제제(NRTI 및 NtRTI)는 이러한 역전사 효소의 기능을 방해한다. 이들 억제제가 효과를 나타내려면 먼저 세포 안으로 들어가 활성화되어야 한다. 세포 내 유입은 주로 수동 확산이나 담체 매개 수송(carrier-mediated transport)을 통해 이루어진다. NRTI는 대체로 친수성이어서 세포막 투과성이 제한적이므로 이 단계가 중요하다.[35]
NRTI와 NtRTI는 내인성 2'-디옥시뉴클레오사이드 및 뉴클레오타이드와 유사한 구조를 가진다. 이들은 처음에는 비활성 상태(모체 원형)이므로, 세포 내에서 여러 단계의 인산화 과정을 거쳐 활성 형태로 전환되어야 한다.[35]
이러한 단계적 활성화 과정은 세포 내 여러 효소들의 연쇄 반응을 통해 일어난다.[40] NRTI의 첫 번째 인산화는 주로 디옥시뉴클레오사이드 키네이스(kinase)에 의해 촉매되며, 이 과정이 전체 활성화 속도를 결정하는 속도 결정 단계인 경우가 많다. 두 번째 인산화는 주로 뉴클레오사이드 일인산 키네이스(NMP kinase)가, 마지막 세 번째 인산화는 뉴클레오사이드 이인산 키네이스(NDP kinase), 포스포글리세르산 키네이스(phosphoglycerate kinase), 피루브산 키네이스, 크레아틴 키네이스(creatine kinase) 등 다양한 효소가 촉매할 수 있다. 이 과정을 통해 NRTI/NtRTI는 항바이러스 활성을 갖는 삼인산 형태로 전환된다.[35]
활성화된 삼인산 형태의 NRTI/NtRTI는 자연적인 디옥시뉴클레오사이드 삼인산(dNTP)과 경쟁하여, 역전사 효소에 의해 새로 만들어지는 바이러스 DNA 사슬에 끼어들어간다(그림 1 참조).[35] 하지만 자연적인 dNTP와 달리, NRTI/NtRTI는 디옥시리보스의 3' 위치에 하이드록시기(-OH)가 없다. 이 3'-수산기는 다음 뉴클레오타이드와의 인산다이에스터 결합 형성에 필수적이므로, NRTI/NtRTI가 DNA 사슬에 삽입되면 더 이상 DNA 사슬이 길어지지 못하고 합성이 중단된다. 즉, 이들 억제제는 DNA 사슬 종결자(chain terminator)로 작용하여 바이러스 복제를 막는다.[39][41]
3. 2. 구조
HIV-1 역전사효소는 비대칭적인 헤테로다이머(heterodimer)로, 총 1000개의 아미노산으로 이루어져 있으며 두 개의 단백질 소단위체(subunit)로 구성된다.[8][37] 이 두 소단위체는 크기와 역할이 다르다.
4. 뉴클레오사이드/뉴클레오타이드 역전사 효소 억제제 (NRTI/NtRTI) 작용 기전
뉴클레오사이드 역전사 효소 억제제(NRTI)와 뉴클레오타이드 역전사 효소 억제제(NtRTI)는 세포 내로 들어가기 위해 주로 수동 확산이나 운반체 매개 수송 방식을 이용한다. NRTI는 친수성이 높아 세포막 투과성이 제한적이므로 이 세포 진입 단계가 중요하다.[35][6]
NRTI와 NtRTI는 체내에 존재하는 2'-디옥시뉴클레오사이드와 뉴클레오타이드의 유사체이다. 이들은 세포에 들어온 초기 형태로는 비활성 상태이며, 항바이러스 효과를 나타내기 위해 연속적인 인산화 과정을 거쳐 활성화되어야 한다.[35][6] 뉴클레오사이드(NRTI)는 총 세 번의 인산화가 필요하고, 이미 인산기를 하나 가지고 있는 뉴클레오타이드(NtRTI)는 두 번의 인산화만 거치면 된다.[39][10]
이 단계적인 활성화 과정은 세포 내에서 여러 효소들의 정교한 연쇄 반응을 통해 이루어진다.[40][11] NRTI의 첫 번째 인산화는 전체 반응 속도를 결정하는 속도 결정 단계인 경우가 많으며, 주로 디옥시뉴클레오사이드 키네이스에 의해 촉매된다. 두 번째 인산기는 일인산 뉴클레오사이드 인산화효소(NMP kinase)가 붙여준다. 마지막 인산화 단계는 이인산 뉴클레오사이드 인산화효소(NDP kinase), 포스포글리세레이트 키나제, 피루브산 키네이스, 크레아틴 키네이스 등 다양한 효소들에 의해 촉매되어, 최종적으로 항바이러스 활성을 갖는 삼인산 유사체 형태로 전환된다.[35][6]
활성화된 삼인산 형태의 NRTI/NtRTI는 바이러스의 역전사 효소가 새로운 DNA 가닥을 합성할 때 필요한 재료인 내인성 디옥시뉴클레오사이드 삼인산(dNTP)과 경쟁하여 DNA 사슬에 삽입된다.[35][6] 하지만 일반적인 dNTP와 달리, NRTI/NtRTI는 디옥시리보스의 3번 탄소 위치에 수산기(-OH)가 없다.[39][10][12] 이 3'-수산기는 DNA 사슬이 계속 길어지기 위해 다음 핵산과 5'→3' 방향으로 인산다이에스터 결합을 형성하는 데 필수적이다.[39][41][10][12] 따라서 NRTI/NtRTI가 일단 DNA 사슬에 삽입되면, 3'-수산기의 부재로 인해 역전사 효소는 더 이상 DNA 사슬을 연장할 수 없게 된다. 즉, NRTI/NtRTI는 DNA 합성을 중단시키는 사슬 종결자(chain terminator) 역할을 함으로써 바이러스 증식을 억제한다.[39][41][10][12] 예를 들어, 최초의 NRTI 중 하나인 지도부딘(AZT)은 원래의 뉴클레오사이드인 티미딘의 3' 수산기 대신 아자이드기가 붙어 있어, DNA 사슬에 결합하면 더 이상의 사슬 연장을 불가능하게 만든다.[13][14]
5. 뉴클레오사이드/뉴클레오타이드 역전사 효소 억제제 (NRTI/NtRTI) 발견과 발전
1981년 여름 후천성 면역 결핍 증후군(AIDS)이 처음 보고되었고,[1] 2년 뒤 AIDS의 병인으로 인체 면역 결핍 바이러스(HIV)가 확인되었다.[2][3] HIV 발견 이후, 효과적인 항레트로바이러스 약물 개발과 HIV 연구는 큰 진전을 이루었다.[3][4]
HIV 감염 치료를 위한 항레트로바이러스 약물은 크게 6가지 종류로 나뉜다. 이 중 HIV-1의 역전사효소를 표적으로 하는 뉴클레오사이드 및 뉴클레오타이드 역전사 효소 억제제(NRTI 및 NtRTI)는 항 HIV 약물 개발의 중요한 기반이 되었다.[5] 최초로 시험관 내에서 항 HIV 활성을 보인 NRTI는 지도부딘이었다.[6]
지도부딘이 1987년에 승인된 이후, 여러 NRTI와 하나의 뉴클레오타이드 역전사 효소 억제제(NtRTI)가 미국 식품의약국(FDA)의 승인을 받았다.[6] FDA 승인을 받은 주요 NRTI/NtRTI는 다음과 같다.[4][6]
| 약물명 | 구분 |
|---|---|
| 지도부딘 | NRTI |
| 디다노신 | NRTI |
| 잘시타빈 | NRTI |
| 스타부딘 | NRTI |
| 라미부딘 | NRTI |
| 아바카비르 | NRTI |
| 엠트리시타빈 | NRTI |
| 테노포비르 | NtRTI |
NRTI와 NtRTI는 우리 몸에 원래 존재하는 내생적 2´-데옥시뉴클레오사이드 및 뉴클레오타이드와 유사한 구조를 가진다. 이 약물들은 세포 안으로 들어간 뒤 여러 단계의 인산화 과정을 거쳐 활성 형태로 전환되어야 항바이러스 효과를 나타낸다.[6][10] 활성화된 약물은 HIV 역전사효소가 바이러스의 유전 물질인 DNA를 합성하는 과정에 끼어들어, DNA 사슬이 더 이상 길어지지 못하게 막는 '사슬 종결자' 역할을 함으로써 바이러스 증식을 억제한다.[10][12] 각 약물의 구체적인 개발 과정과 작용 방식은 이어지는 하위 섹션에서 자세히 설명한다.
5. 1. HIV 치료를 향한 첫걸음 - 지도부딘
1964년, 미국 미시간 암 재단의 호르위츠(Horwitz) 연구팀은 지도부딘(AZT)이라는 화합물을 처음 합성했다.[42] 지도부딘은 DNA 구성 요소 중 하나인 티민과 유사한 구조를 가지지만, 디옥시리보스 고리의 특정 위치(3' 탄소)에 수산화기(-OH) 대신 아지도기(-N3)가 붙어 있는 점이 다르다.[42]DNA가 복제될 때는 효소(역전사효소)가 새로운 뉴클레오타이드를 하나씩 붙여나가는데, 이때 3' 수산화기가 다음 뉴클레오타이드가 결합하는 연결점 역할을 한다. 지도부딘에는 이 3' 수산화기가 없기 때문에, HIV의 유전 물질이 복제되는 과정에서 지도부딘이 DNA 사슬에 끼어들어가면 더 이상 사슬이 길어지지 못하고 복제가 중단된다. 즉, 지도부딘은 '사슬 종결자'로 작용하여 HIV의 증식을 강력하게 억제한다.[42][43]
처음 지도부딘이 합성되었을 때는 암 치료를 위한 항암제 후보 물질로 연구되었으나, 암세포에 대한 효과는 없는 것으로 밝혀졌다.[42] 이후 1974년에 지도부딘이 레트로바이러스에 효과가 있다는 연구 결과가 보고되었고, 1980년대 중반 에이즈(AIDS)가 전 세계적으로 유행하면서 HIV 치료제로서 다시 주목받게 되었다.[42][44]
하지만 지도부딘은 부작용 문제도 가지고 있었다. 지도부딘은 바이러스에 감염된 세포뿐만 아니라 감염되지 않은 정상 세포 안에서도 세포 내 효소에 의해 활성 형태(삼인산 형태)로 전환될 수 있어, 정상 세포에도 독성을 나타낼 수 있다는 단점이 있다.[43]
5. 2. 뉴클레오사이드 유사체들의 발전
뉴클레오사이드 및 뉴클레오타이드 역전사 효소 억제제(NRTI 및 NtRTI)의 활성화는 주로 수동 확산 또는 운반체 매개 수송을 통해 세포 안으로 들어가는 것에 의존한다. NRTI는 매우 친수성이라서 막을 통과하기 어렵기 때문에 이 단계가 중요하다.NRTI는 우리 몸에 원래 존재하는 내생적 2´-데옥시뉴클레오사이드 및 뉴클레오타이드와 유사한 구조를 가진다. NRTI는 처음 약물 형태 그대로는 효과가 없으며, 세포 내에서 연속적인 인산화 과정을 거쳐 활성화되어야 한다.[6]
뉴클레오사이드 유사체는 세 번의 인산화 과정을 거쳐야 활성화되는 반면, 인산기를 이미 하나 가지고 있는 뉴클레오타이드 유사체는 두 번의 인산화만 거치면 된다.[10] 인산화 단계가 적다는 것은 약물이 활성 형태(대사 산물)로 더 빠르고 완전하게 전환될 수 있음을 의미한다.[6] 이러한 단계별 활성화 과정은 세포 내부에서 일어나며, 여러 효소들이 순서대로 작용하여 진행된다.[11] 첫 번째 인산화(뉴클레오사이드 유사체의 경우 종종 전체 반응 속도를 결정하는 속도 제한 단계)는 주로 데옥시뉴클레오사이드 키나제에 의해 촉매된다. 뉴클레오사이드 일인산 유사체에 두 번째 인산기를 붙이는 것은 뉴클레오사이드 일인산 키나제(NMP 키나제)가 담당한다. 마지막 인산화 단계는 뉴클레오사이드 이인산 키나제(NDP 키나제), 포스포글리세레이트 키나제, 피루브산 키나제, 크레아틴 키나제 등 다양한 효소들이 촉매하여 항바이러스 활성을 가진 삼인산 형태로 만든다.[6]
각각의 삼인산 형태로 활성화된 NRTI와 유일한 NtRTI는 HIV 역전사효소(RT)가 새로운 DNA 가닥을 만들 때, 우리 몸의 정상적인 재료인 데옥시뉴클레오타이드 삼인산(dNTP)과 경쟁하여 자신이 대신 DNA 가닥에 끼어 들어간다.[6] 하지만 dNTP와 달리 NRTI/NtRTI는 데옥시리보스 부분에 다음 핵산과 연결되는 데 필요한 3´-수산기(-OH)가 없다. 일단 DNA 가닥에 삽입되면, 이 3´-수산기가 없기 때문에 역전사효소는 더 이상 DNA 가닥을 길게 만들 수 없게 된다. 즉, NRTI/NtRTI는 DNA 합성을 중간에 멈추게 하는 사슬 종결자 역할을 한다.[10][12] 이러한 방식으로 HIV의 증식을 억제한다. 구체적인 약물들의 개발 과정은 이어지는 하위 섹션들에서 자세히 설명한다.
5. 2. 1. 다이디옥시뉴클레오사이드(dideoxynucleoside)
wikitext| 화학 물질 | 화학 구조 |
|---|---|
| 다이디옥시아데노신 |  |
| 다이다노신 |  |
다이디옥시뉴클레오사이드(dideoxynucleoside)는 당 고리에 2'-수산화기(hydroxyl group)와 3'-수산화기가 모두 없는 뉴클레오사이드 유사체이다.[38] 지도부딘이 합성된 지 3년 후, 시카고의 제롬 호르위츠(Jerome Horwitz)와 그의 동료들은 현재 잘시타빈(Zalcitabine, ddC)으로 알려진 또 다른 다이디옥시뉴클레오사이드를 개발했다.[45][16] 잘시타빈은 합성된 피리미딘 뉴클레오사이드 유사체로, 데옥시시티딘과 구조적으로 관련이 있으며 리보스 당의 3'-수산화기가 수소로 치환되어 있다.[46][17] 잘시타빈은 1992년 6월 FDA으로부터 HIV-1 치료 효과를 인정받아 승인되었다.[32][47][3][18]
2',3'-다이디옥시이노신(dideoxyinosine) 또는 다이다노신(Didanosine, ddI)은 체내에서 다이디옥시아데노신(dideoxyadenosine)으로 전환되는 약물로, 개발 역사가 길다.[48][19] 1964년에 잘시타빈에 상응하는 아데노신 유사체인 다이디옥시아데노신이 합성되었으나, 신장 손상을 유발하는 문제가 있었다. 이 때문에 다이디옥시아데노신을 효소적 산화(enzymatic oxidation)시켜 다이다노신을 만들게 되었다(표 1 참조). 다이다노신은 신장 손상 없이 HIV에 효과적인 것으로 밝혀졌다.[45][16] 다이다노신은 1991년 10월 FDA로부터 HIV-1 치료제로 승인받았다.[47][9]
잘시타빈과 다이다노신은 모두 DNA 사슬 합성을 중단시키는 사슬 종결자(chain terminator)로서 항-HIV 치료제로 개발되었다. 그러나 두 약물 모두 선택성이 낮아 부작용을 일으키는 단점이 있다.[43]
5. 2. 2. 추가 개발
디데옥시 구조의 추가적인 변형을 통해 2´,3´-디데히드로-3´-데옥시티미딘(스타부딘, d4T)이 개발되었다. 스타부딘의 활성은 지도부딘의 활성과 유사한 것으로 나타났지만, 인산화 패턴은 다르다. 즉, 티미딘 키나아제(첫 번째 인산화를 담당하는 효소)에 대한 지도부딘의 친화력(약리학)은 티미딘과 유사한 반면, 스타부딘의 친화력은 700배 더 약하다.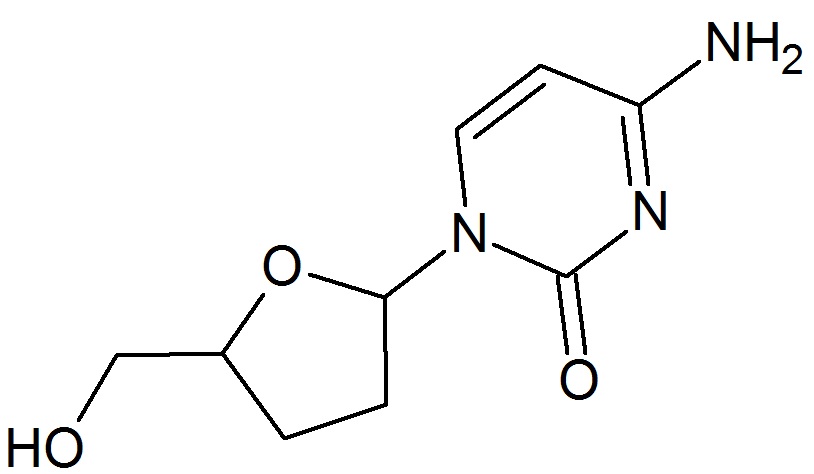

2',3'-디데옥시-3'-티아시티딘(라미부딘, 3TC)은 버나드 벨로에 의해 발견되었다. 라미부딘의 역사는 1970년대 중반, 버나드 벨로가 당 유도체(화학)를 연구하던 시기로 거슬러 올라간다. 라미부딘은 잘시타빈의 황 유사체로 개발되었다(표 2 참조).[16] 처음에는 라세미 혼합물(BCH-189)로 합성되었으며, 분석 결과 BCH-189(2',3'-디데옥시-3'-티아시티딘)의 양성 및 음성 에난티오머 모두 HIV에 대해 체외 활성을 나타냈다. 라미부딘은 음성 에난티오머이며 피리미딘 뉴클레오사이드 유사체이다. 2'-데옥시시티딘의 리보스 고리 3' 탄소는 황 원자로 대체되었는데, 이는 양성 에난티오머보다 항 HIV 활성이 더 크고 독성이 적기 때문이다.[16][20][21]
다음으로 2',3'-디데옥시-5-플루오로-3'-티아시티딘(엠트리시타빈, FTC)이 등장했는데, 이는 라미부딘의 구조적 상동계열이다. 구조적 차이점은 라미부딘의 염기 부분에 5-플루오로 변형이 있다는 것이다. 엠트리시타빈은 라미부딘과 여러 면에서 유사하며 HIV-1과 B형 간염 바이러스 모두에 대해 활성이 있다.[21][29]
5. 2. 3. 탄소고리 뉴클레오사이드
다이디옥시아데노신의 탄소고리 유사체들은 항-HIV 효과가 있는지 연구되었다.[16] 처음에는 미미한 활동만이 관찰되었다. 많은 뉴클레오사이드 유사체들이 준비되고 검토되었지만, 오직 한 가지만이 눈에 띄는 활동을 보였고 임상시험 사용 기준을 만족했다. 그것은 다이디옥시아데노신의 2',3'-다이디하이드로 유사체였다.[16] 아데닌 고리의 6-아미노기 질소에 사이클로프로필기(cyclopropyl)를 추가하여 화합물의 지질친화도(lipophilicity)를 높였고, 이를 통해 뇌 침투율을 향상시켰다. 그 결과 만들어진 화합물이 아바카비르(abacavir)이다 (표 3 참조).[16] 아바카비르는 1998년 12월 미국 식품의약국(FDA)으로부터 HIV-1 감염 치료제로 승인받았다.[20]아바카비르는 체내에서 활성을 나타내는 유일하게 승인된 구아노신 유사체 항레트로바이러스제이다.[9] 이 약물은 먼저 아데노신 포스포트랜스퍼라제에 의해 일인산화된 후, 생성된 일인산 화합물이 카보비르 3'-일인산(carbovir 3'-monophosphate)으로 전환된다. 이후 완전히 인산화된 카보비르는 역전사효소(RT)에 의해 DNA 사슬에 삽입되어 사슬 종결자 역할을 한다.[9] 관련된 구아노신 유도체인 카보비르(carbovir)는 구강 생체이용률이 낮아 임상 개발이 중단되었다.[9]
| 디데옥시아데노신 | 다이다노신 | 아바카비르 | |
|---|---|---|---|
| 화학 구조 |  | -- |  |
5. 2. 4. 비고리형 뉴클레오타이드 - 승인된 유일한 뉴클레오타이드 역전사효소 억제제
뉴클레오사이드 유사체는 활성화되기 위해 세 번의 인산화 과정을 거쳐야 하지만, 뉴클레오타이드 유사체는 이미 인산기를 하나 가지고 있어 두 번의 인산화만 필요하다.[10] 이렇게 인산화 단계가 줄어들면 약물이 활성 대사 산물로 더 빠르고 완전하게 전환될 수 있다는 장점이 있다.[6] 이러한 점을 고려하여 테노포비르(Tenofovir)와 같은 포스폰산 뉴클레오타이드 유사체가 개발되었다.테노포비르는 비고리형(acyclic) 아데노신 유도체로, 포스폰산 부분을 가지고 있다는 점이 특징이다. 이러한 비고리형 구조와 포스폰산 부분은 미국 식품의약국(FDA)에서 승인한 뉴클레오타이드 역전사효소 억제제(NtRTI) 중에서는 테노포비르만이 가진 독특한 구조적 특징이다.[21]
실제 약물로 사용되는 것은 테노포비르의 전구약물인 테노포비르 디소프록실 푸마르산염(Tenofovir Disoproxil Fumarate, TDF)이다. 테노포비르 DF는 체내에서 효소에 의해 가수분해되어 항-HIV 활성을 나타내는 테노포비르로 전환된다.[22][23] 이 약물은 2,3-디하이드록시프로필아데닌이라는 물질의 합성과 광범위한 항바이러스 활성 연구를 통해 개발되었다.[23]
테노포비르 DF는 2001년 10월, HIV-1 감염 치료제로 FDA의 승인을 받으며 최초의 뉴클레오타이드 역전사효소 억제제가 되었다.[9][22] 현재까지 승인된 유일한 뉴클레오타이드 역전사효소 억제제이기도 하다.[4][6]
6. 뉴클레오사이드/뉴클레오타이드 역전사 효소 억제제 (NRTI/NtRTI) 저항성
HIV-1이 항바이러스 치료제에 지속적으로 노출되면서 약물 내성 바이러스가 나타나는 것은 피할 수 없는 결과이다.[54][24] 약물 내성은 바이러스 감염 치료, 특히 HIV 치료에서 심각한 임상적 문제로 여겨진다.[54][24] 현재까지 승인된 모든 뉴클레오사이드 역전사효소 억제제(NRTI)에서 저항성 돌연변이가 관찰되었다.[55][25]
NRTI에 대한 약물 내성이 생기는 주요 기전은 두 가지로 밝혀졌다: NRTI가 DNA에 삽입되는 것을 방해하거나, 이미 삽입된 NRTI를 제거하는 것이다.[55][56][25][26]
첫 번째 기전인 NRTI 삽입 방해는 역전사효소(RT)의 p66 서브도메인에서 돌연변이가 발생하여 일어난다.[56][26] 이 돌연변이는 입체 장애를 유발하여 특정 약물(예: 라미부딘)이 효소의 활성 부위에 결합하여 DNA 사슬에 삽입되는 것을 막는다.[56][26]
두 번째 기전은 이미 삽입된 NRTI를 제거하는 것이다. 저항성을 가진 RT 효소는 NRTI를 DNA 사슬에 일단 삽입한 후, 중합반응 단계를 역전시켜 삽입된 NRTI를 제거할 수 있다.[56][26] 이 제거 과정에는 파이로인산염(pyrophosphate) 제공자가 필요하며, RT는 NRTI의 3' 프라이머 말단에 파이로인산염을 결합시켜 프라이머 DNA로부터 떼어낸다.[56][26]
환자에게서 HIV-1 복제를 효과적으로 억제하고 약물 내성 바이러스의 출현을 최대한 늦추기 위해 여러 약물을 함께 사용하는 병용 요법이 사용된다.[57][27] HAART(고강도 항레트로바이러스 치료법)는 NRTI, 뉴클레오타이드 역전사효소 억제제(NtRTI), 비뉴클레오사이드 역전사효소 억제제(NNRTI), 단백질분해효소 억제제 등 다양한 종류의 항바이러스 약물을 조합하여 사용한다.[57][27]
7. 현재 개발 중인 뉴클레오사이드/뉴클레오타이드 역전사 효소 억제제 (NRTI/NtRTI)
최근 여러 뉴클레오사이드 역전사효소 억제제(NRTI) 및 뉴클레오타이드 역전사효소 억제제(NtRTI)가 임상시험부터 전임상 개발까지 다양한 단계에서 연구되고 있으며, 이는 기존 약물의 독성을 줄이고 내성 바이러스에 대한 효과를 높이며 HIV-1 치료를 단순화하기 위함이다.[35][55][58][6][25][28]
이 외에도 여러 NRTI/NtRTI 후보 물질들이 미국 식품의약국(FDA)의 임상시험계획(IND, Investigational New Drug) 승인을 받았거나 다양한 임상 단계에 있으며, 이들은 새로운 치료제가 필요한 환자들에게 유용할 수 있는 약리학적 특징을 보여주고 있다.[35][51][58][6][29][9]
7. 1. 개발 중인 약물 후보
최근 여러 뉴클레오사이드 역전사효소 억제제(NRTI)가 임상시험에서부터 전임상 개발 단계에 이르기까지 다양한 단계에서 개발되고 있다. 새로운 NRTI를 지속적으로 연구하는 주된 이유는 기존 약물의 독성을 줄이고, 내성을 보이는 바이러스에 대한 효과를 높이며, HIV-1 감염 치료 과정을 단순화하기 위해서이다.[6][25][28][35][55][58]- '''아프리시타빈''' (Apricitabine, ATC): 데옥시시티딘 유사체로, 라미부딘과 구조적으로 관련이 있으며 분자 내 산소와 황의 위치가 바뀌어 있다.[21][50] 다른 NRTI에 비해 시험관 내에서의 활성은 다소 낮지만, NRTI에 내성을 가진 다양한 HIV-1 변종에 대해서도 효과를 유지한다는 장점이 있다. 아프리시타빈은 이전에 NRTI 치료 경험이 있는 환자들을 대상으로 한 임상 개발의 마지막 단계에 있다.[6][35]
- '''엘부시타빈''' (Elvucitabine, L-d4FC): 데옥시시티딘 유사체이며, 지도부딘이나 라미부딘 등 여러 뉴클레오사이드 유사체에 내성을 보이는 HIV에 대해서도 효과가 있다.[29][51] 이는 세포 내에서 약물의 활성 형태인 삼인산 대사 산물 농도가 매우 높게 유지될 수 있기 때문이다.[6][35] 그러나 일부 환자에게서 골수 억제 부작용이 나타났고, 약물 투여 시작 후 이르면 이틀 만에 CD4+ 세포 수치가 감소하는 현상이 관찰되어 현재 임상 시험이 보류된 상태이다.[9][29][51][58]
- '''암독소비르''' (Amdoxovir, DAPD): 생체이용률이 좋은 구아노신 유사체 NRTI 프로드러그이다.[6][29][9][35][51][58] 세포 내에서 아데노신 탈아미노효소에 의해 디옥솔란 구아닌(DXG)으로 변환된다. 약물의 실제 활성 형태인 DXG-삼인산은 기존 형태인 DAPD-삼인산보다 더 강력한 효과를 나타낸다.[29][51] 암독소비르는 현재 2상 임상 시험 단계에 있다.[9][23][53][58]
- '''라시비르''' (Racivir, RCV): 엠트리시타빈(FTC)의 두 가지 광학 이성질체인 (-)-FTC와 (+)-FTC가 1:1로 섞인 라세미 혼합물이다. 라시비르는 경구 생체이용률이 우수하며 하루에 한 번만 복용하면 된다는 장점이 있다. 다른 NRTI와 병용 투여했을 때 유망한 항바이러스 효과를 보였으며, 현재 2상 임상 시험이 진행 중이다.[6][29][9]
| 약물 후보 | 아프리시타빈 | 엘부시타빈 | 암독소비르 | 라시비르 |
|---|---|---|---|---|
| 화학 구조 |  | 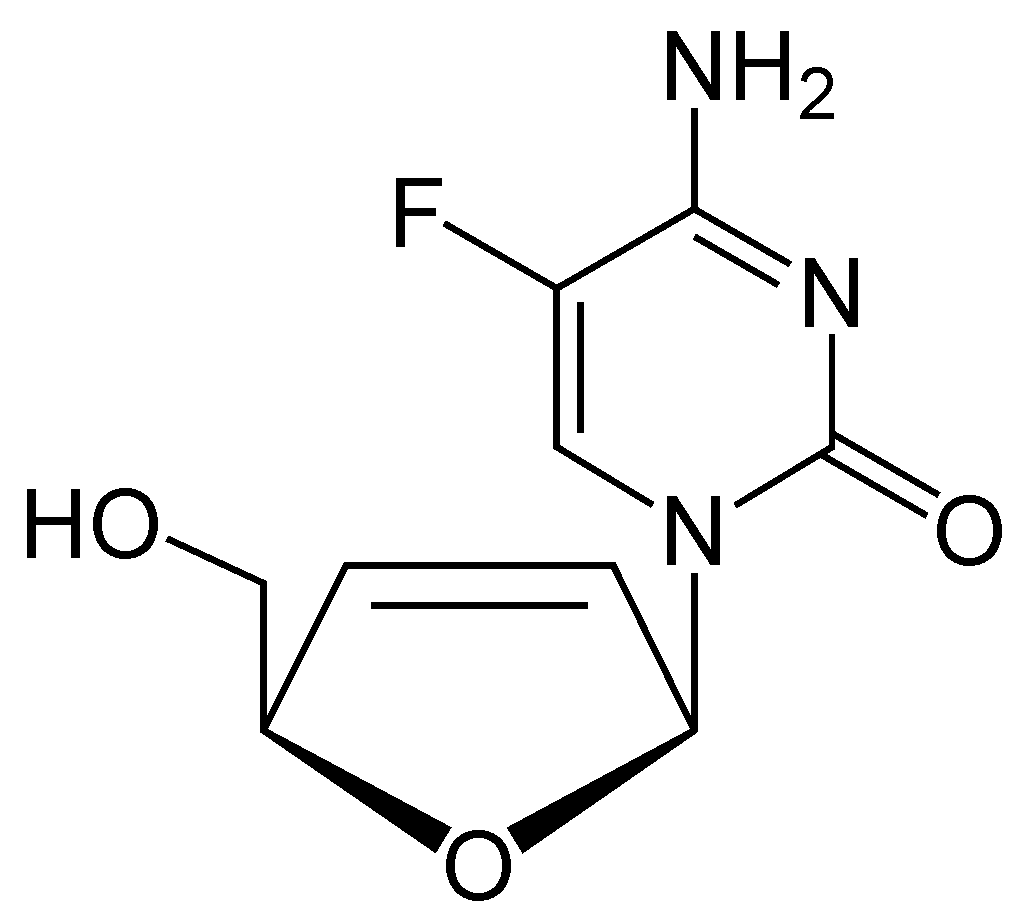 | 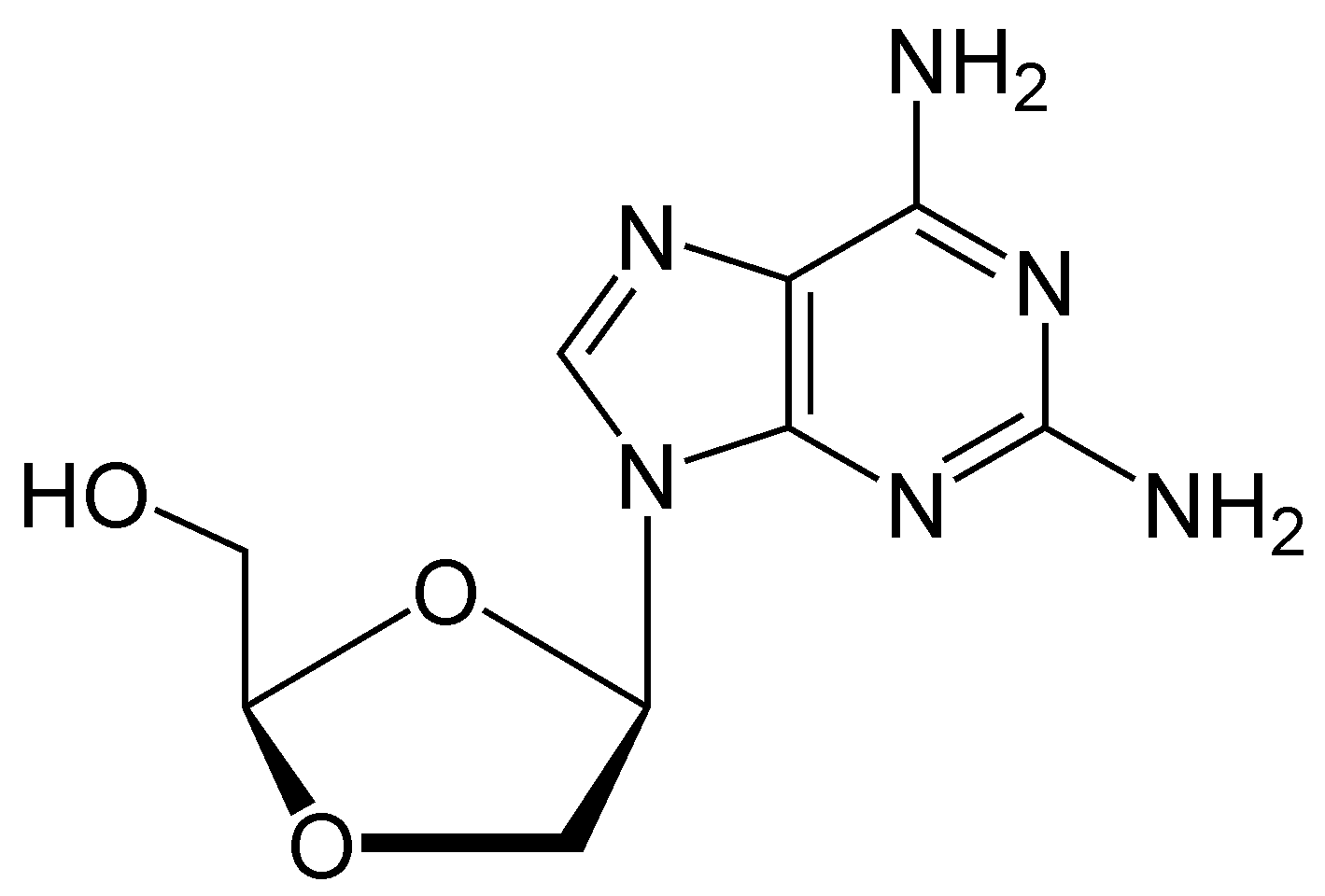 |  |
| 개발 단계 | 임상 개발 마지막 단계 | 중단됨 | 2상 | 2상 |
이 외에도 더 많은 NRTI들이 개발 중에 있다. 개발사들이 미국 식품의약국(FDA)에 임상시험계획(IND) 신청서를 제출했거나 승인을 받았으며, 여러 임상 시험 단계에 있는 약물들이 있다. 개발 중인 일부 NRTI는 새로운 치료제가 필요한 환자들에게 유용할 수 있는 다양한 약리학적 특성을 보여주고 있다.[6][29][9]
8. 같이 보기
- 항레트로바이러스 의약품
- CCR5 수용체 길항제의 발견과 발전
- 비뉴클레오사이드 역전사효소 억제제의 발견과 발전
- HIV 단백질분해효소 억제자의 발견과 발전
- 역전사효소 억제제
- 단백질분해효소 억제제
- 진입 억제제
참조
[1]
논문
HIV–AIDS Pandemic at 25 — The Global Response
[2]
논문
The AIDS epidemic Considerations for the 21st Century
[3]
논문
HIV and AIDS: 20 years of science
https://zenodo.org/r[...]
[4]
논문
Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV
[5]
논문
HIV-1 reverse transcriptase connection subdomain mutations reduce template RNA degradation and enhance AZT excision
[6]
논문
Nucleoside and nucleotide HIV reverse-transcriptase inhibitors: 25 years after zidovudine
[7]
논문
Retroviral reverse transcriptases (other than those of HIV-1 and murine leukemia virus): A comparison of their molecular and biochemical properties
[8]
논문
The search for potent, small molecule NNRTIs: A review
[9]
논문
Reverse transcription of the HIV-1 pandemic
[10]
논문
Primer unblocking by HIV-1 reverse transcritptase and resistance to nucleoside RT inhibitors
[11]
논문
Pharmacology of nucleoside and nucleotide Reverse transcriptase inhibitor - induced mitochondrial toxicity
[12]
논문
Retroviral reverse transcriptases
[13]
서적
Drug prototypes and their exploitation
John Wileys & sons
[14]
서적
Smith and Williams´ Introduction to the principles of drug design and action
Harwood academic publishers
[15]
간행물
Top drugs: Top synthetic routes
[16]
서적
Drug discovery a history
https://books.google[...]
Wiley
[17]
간행물
National institute of allergy and infectious diseases, NIH
https://books.google[...]
[18]
논문
Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV
[19]
서적
Goodman & Gilman's The Pharmacological Basis of Therapeutics, Eleventh Edition
McGraw-Hill
[20]
서적
Reverse transcriptase inhibitors in HIV/AIDS therapy
Humana press inc
[21]
간행물
Antiviral research strategies in antiviral drug discovery
AMS press
[22]
논문
Tenofovir Disoproxil Fumarate: A Nucleotide ReverseTranscriptase Inhibitor for the Treatment of HIV Infection
[23]
논문
Development of novel nucleoside analogues for use against drug resistant strains of HIV-1
[24]
논문
Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors
http://biomaps.rutge[...]
2010-11-03
[25]
논문
The "Connection" Between HIV Drug Resistance and RNase H
[26]
논문
Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase
[27]
논문
Mechanism of Action and In Vitro Activity of 1',3'-Dioxolanylpurine Nucleoside Analogues against Sensitive and Drug-Resistant Human Immunodeficiency Virus Type 1 Variants
[28]
논문
An overview on HIV-1 reverse transcriptase inhibitors
http://www.chalcogen[...]
[29]
논문
New nucleoside reverse transcriptase inhibitors for the treatment of HIV infections
[30]
논문
HIV–AIDS Pandemic at 25 — The Global Response
[31]
논문
The AIDS epidemic Considerations for the 21st Century
[32]
논문
HIV and AIDS: 20 years of science
https://zenodo.org/r[...]
[33]
논문
Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV
[34]
논문
HIV-1 reverse transcriptase connection subdomain mutations reduce template RNA degradation and enhance AZT excision
[35]
논문
Nucleoside and nucleotide HIV reverse-transcriptase inhibitors: 25 years after zidovudine
[36]
논문
Retroviral reverse transcriptases (other than those of HIV-1 and murine leukemia virus): A comparison of their molecular and biochemical properties
[37]
논문
The search for potent, small molecule NNRTIs: A review
[38]
논문
Reverse transcription of the HIV-1 pandemic
[39]
논문
Primer unblocking by HIV-1 reverse transcritptase and resistance to nucleoside RT inhibitors
[40]
논문
Pharmacology of nucleoside and nucleotide Reverse transcriptase inhibitor - induced mitochondrial toxicity
[41]
논문
Retroviral reverse transcriptases
[42]
서적
Drug prototypes and their exploitation
John Wileys & sons
[43]
서적
Smith and Williams´ Introduction to the principles of drug design and action
Harwood academic publishers
[44]
서적
Top drugs: Top synthetic routes
[45]
서적
Drug discovery a history
https://books.google[...]
[46]
서적
National institute of allergy and infectious diseases, NIH
https://books.google[...]
[47]
논문
Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV
[48]
서적
Goodman & Gilman's The Pharmacological Basis of Therapeutics, Eleventh Edition
McGraw-Hill
[49]
서적
Reverse transcriptase inhibitors in HIV/AIDS therapy
Humana press inc
[50]
서적
Antiviral research strategies in antiviral drug discovery
AMS press
[51]
논문
New nucleoside reverse transcriptase inhibitors for the treatment of HIV infections
[52]
논문
Tenofovir Disoproxil Fumarate: A Nucleotide ReverseTranscriptase Inhibitor for the Treatment of HIV Infection
[53]
논문
Development of novel nucleoside analogues for use against drug resistant strains of HIV-1
[54]
논문
Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors
http://biomaps.rutge[...]
2021-07-02
[55]
논문
The "Connection" Between HIV Drug Resistance and RNase H
[56]
논문
Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase
[57]
논문
Mechanism of Action and In Vitro Activity of 1',3'-Dioxolanylpurine Nucleoside Analogues against Sensitive and Drug-Resistant Human Immunodeficiency Virus Type 1 Variants
[58]
논문
An overview on HIV-1 reverse transcriptase inhibitors
http://www.chalcogen[...]
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.
문의하기 : help@durumis.com