기능유전체학
"오늘의AI위키"의 AI를 통해 더욱 풍부하고 폭넓은 지식 경험을 누리세요.
1. 개요
기능유전체학은 유전자, 단백질, 게놈 전체 구성 요소의 기능을 연구하여 생물체의 동적 특성을 이해하는 학문이다. 유전체학 및 단백질체학 지식을 통합하여 게놈과 표현형 간의 관계를 밝히는 것을 목표로 하며, 유전자 변이가 질병으로 이어지는 과정을 이해하는 데 기여한다. 기능유전체학 연구는 DNA, RNA, 단백질 수준에서의 다양한 기술을 활용하며, 유전자 상호작용 매핑, DNA/단백질 상호작용 분석, RNA 시퀀싱, 질량 분석법 등을 통해 유전자 기능과 상호 작용을 연구한다. 또한, ENCODE, GTEx, AVE와 같은 국제 컨소시엄 프로젝트를 통해 인간 유전체 연구를 진행하고 있다.

더 읽어볼만한 페이지
- 체학 - 화학유전체학
화학유전체학은 활성 화합물을 탐침으로 사용하여 단백질체 기능 특성화를 수행하고 표적과 약물 발견을 통합하는 전략으로, 저분자 화합물과 단백질 간 상호 작용으로 유도되는 표현형 분석을 통해 단백질과 분자적 사건 간의 연결을 밝히며 신약 개발 등 다양한 분야에 응용된다. - 체학 - 글리콤
글리콤 연구에는 다중 반응 모니터링, 렉틴 및 항체 어레이, 고분해능 질량 분석법, 고성능 액체 크로마토그래피 등 다양한 기술이 사용되며, 이 문서에서는 글리칸 대사 연구 및 당단백질 분석에 사용되는 도구와 질량 분석법의 장단점을 설명한다. - 유전체학 - 발현체학
- 유전체학 - 유전형 분석
유전자형 분석은 DNA 염기 서열 분석, RFLP, PCR, DNA 마이크로어레이 등 다양한 기술을 통해 유전형을 파악하는 방법으로 질병 진단, 품종 개량 등 여러 분야에 활용되며 윤리적 문제와 사회적 영향을 동반하지만, 혁신적인 변화를 가져올 것으로 예상되는 기술이다. - 분자생물학 - 단백질
단백질은 아미노산 중합체로 생체 구조 유지와 기능에 필수적이며, 아미노산 서열에 따라 고유한 3차원 구조를 형성하여 효소, 구조, 수송, 저장, 수축, 방어, 조절 단백질 등 다양한 기능을 수행하고, 인체 내에서 건강 유지와 질병 예방에 중요한 역할을 하는 필수 영양소이다. - 분자생물학 - 의학
의학은 질병의 진단, 예후, 치료, 예방을 연구하는 과학 및 실천 분야이며, 고대부터 발전하여 현대에는 다양한 전문 분야로 세분화되고 첨단 기술 발전에 따라 혁신적인 변화를 겪고 있다.
2. 기능유전체학의 정의 및 목표
기능유전체학은 유전자, 단백질, 더 나아가 게놈의 모든 구성 요소의 기능을 이해하는 것을 목표로 한다. 이 용어는 유기체의 유전자와 단백질을 연구하는 여러 기술적 접근 방식을 포괄하며, 일부 학자들은 비유전자 요소를 연구에 포함시키기도 한다.[3] 기능유전체학은 유기체의 발달(시간)이나 신체 부위(공간)에 따른 자연적인 유전적 변이 연구와 돌연변이와 같은 기능적 붕괴를 포함한다.
기능유전체학은 유전체 및 단백체 지식을 통합하여 유기체의 동적 특성을 이해하고, 게놈이 어떻게 기능을 나타내는지에 대한 전체적인 그림을 제공한다. 기능유전체학 데이터의 통합은 시스템 생물학 접근 방식의 일부이다.
기능유전체학은 생물의 게놈과 표현형 간의 관계를 이해하고, 유전자 및 유전자 산물의 전체적인 특성 및 기능을 이해하기 위한 접근 방식을 의미한다.
기능유전체학에 대한 기대는 커지고 있으며, 유전체학 및 단백질체학 지식을 융합하여 세포에서 생체 수준까지 생물의 특성에 대한 이해가 요구된다. 이는 생물의 유전 정보로부터 다양한 생물학적 기능이 어떻게 생겨나는지에 대한 해답으로 이어질 것이다. 특히 인간의 유전 질환에 중요한 의미를 가지는데, 특정 유전자의 변이가 어떻게 유전 질환으로 이어지는지를 이해하면 치료 방향을 알 수 있을 것이다.
2. 1. 기능의 정의
Graur 등은 논문에서[1] 기능을 "선택 효과"와 "인과적 역할" 두 가지 방식으로 정의한다. "선택 효과" 기능은 특성(DNA, RNA, 단백질 등)이 선택되는 기능을 의미한다. "인과적 역할" 기능은 특성이 충분하고 필요한 기능을 의미한다. 기능유전체학은 일반적으로 기능의 "인과적 역할" 정의를 검증한다.3. 기능유전체학의 기술 및 응용
기능유전체학은 돌연변이와 다형성(단일 염기 다형성 (SNP) 분석 등)과 같은 게놈 자체의 기능 관련 측면과 분자 활동의 측정을 포함한다. 후자는 전사체학(유전자 발현), 단백질체학(단백질 생산), 대사체학과 같은 여러 "-오믹스"를 포함한다. 기능유전체학은 주로 다중 기술을 사용하여 생물학적 샘플 내의 많은 또는 모든 유전자 산물(예: mRNA 또는 단백질)의 풍부도를 측정한다. 보다 집중적인 기능유전체학 접근 방식은 한 유전자의 모든 변이의 기능을 테스트하고 활성 판독값으로 시퀀싱을 사용하여 돌연변이의 영향을 정량화할 수 있다. 이러한 측정 양식은 함께 다양한 생물학적 과정을 정량화하고 유전자 및 단백질 기능과 상호 작용에 대한 이해를 개선하기 위해 노력한다.[1]
3. 1. DNA 수준
DNA 수준에서는 유전적 상호작용 매핑, DNA/단백질 상호작용, DNA 접근성 분석 등의 기술이 사용된다.3. 1. 1. 유전적 상호작용 매핑
상위성은 두 유전자가 억제될 때 나타나는 표현형이 단일 유전자 녹아웃의 효과의 합과 다를 수 있다는 것을 의미한다. 즉, 두 가지 다른 유전자 녹아웃의 영향이 가산되지 않을 수 있다.[1]유전자나 유전자 발현의 체계적인 쌍별 삭제 또는 억제를 통해, 물리적으로 상호 작용하지 않더라도 관련된 기능을 가진 유전자를 식별할 수 있다.[1]
3. 1. 2. DNA/단백질 상호작용
단백질은 mRNA(전령 RNA, 단백질 합성을 위한 DNA의 암호화된 정보)의 번역에 의해 형성되며 유전자 발현 조절에 중요한 역할을 한다. 단백질이 유전자 발현을 어떻게 조절하는지 이해하려면 단백질이 상호 작용하는 DNA 서열을 식별해야 한다. DNA-단백질 상호 작용 부위를 식별하기 위해 ChIP-seq, CUT&RUN 시퀀싱 및 Calling Cards 등의 기술이 개발되었다.[4]3. 1. 3. DNA 접근성 분석
ATAC-seq, DNase-Seq, FAIRE-Seq 등의 기술을 사용하여 접근 가능한 게놈 영역을 확인하고 잠재적인 조절 영역을 찾는다.[1]3. 2. RNA 수준
기능유전체학은 전사체학(유전자 발현), 단백질체학(단백질 생성), 대사체학 등 다양한 "오믹스" 분야를 포함한다. 특히, RNA 수준에서 기능유전체학은 생물학적 시료 내의 모든 mRNA 양을 측정하여 다양한 생물 현상을 정량화하고, 유전자 기능과 상호작용에 대한 이해를 높인다.RNA 수준의 연구는 마이크로어레이, 유전자 발현의 연속 분석(SAGE), RNA 시퀀싱, small RNA 시퀀싱, 대규모 병렬 리포터 분석(MPRA), STARR-seq, Perturb-seq 등의 기술을 사용하여 전사체 수준의 변화를 분석한다.[1][5][6][7][8]
3. 2. 1. 마이크로어레이

DNA 마이크로어레이는 주어진 유전자 또는 프로브 DNA 서열에 해당하는 샘플 내 mRNA의 양을 측정한다. 프로브 서열은 고체 표면에 고정되어 형광 표지된 "타겟" mRNA와 혼성화되도록 한다. 각 지점의 형광 강도는 해당 지점에 혼성화된 타겟 서열의 양, 따라서 샘플 내 해당 mRNA 서열의 풍부도에 비례한다. 마이크로어레이는 서로 다른 조건 간의 전사체 수준의 변화와 알려진 기능의 유전자와 공유되는 발현 패턴을 기반으로 주어진 과정에 관련된 후보 유전자를 식별할 수 있게 해준다.
3. 2. 2. SAGE
유전자 발현의 연속 분석(SAGE)은 혼성화가 아닌 RNA 시퀀싱을 기반으로 전사체를 분석하는 방법이다. SAGE는 각 유전자마다 고유한 10~17개 염기쌍 태그의 시퀀싱에 의존한다. 이러한 태그는 폴리아데닐화된 mRNA에서 생성되며 시퀀싱 전에 꼬리-꼬리(end-to-end)로 연결된다. SAGE는 연구할 전사체의 사전 지식에 의존하지 않으므로 세포당 전사체 수에 대한 편향되지 않은 측정을 제공한다.[1]3. 2. 3. RNA 시퀀싱
RNA 시퀀싱은 2016년에 언급된 바와 같이, 최근 몇 년 동안 마이크로어레이와 SAGE 기술을 대체하여 전사체 및 유전자 발현을 연구하는 가장 효율적인 방법이 되었다. 이는 일반적으로 차세대 시퀀싱에 의해 수행된다.[5]시퀀싱된 RNA의 하위 집합은 작은 RNA인데, 이는 전사 및 전사 후 유전자 침묵, 즉 RNA 침묵의 핵심 조절 인자인 비암호화 RNA 분자 종류이다. 차세대 시퀀싱은 비암호화 RNA 발견, 프로파일링 및 발현 분석을 위한 표준 도구이다.
3. 2. 4. Small RNA 시퀀싱
Small RNA 시퀀싱은 마이크로어레이와 SAGE 기술을 대체하여 전사 및 유전자 발현을 연구하는 효과적인 방법으로, 차세대 시퀀싱에 의해 수행된다.[5]Small RNA는 전사 및 전사 후 유전자 침묵, 즉 RNA 침묵의 핵심 조절 인자인 비암호화 RNA 분자이다. 차세대 시퀀싱은 비암호화 RNA 발견, 프로파일링 및 발현 분석을 위한 표준 도구이다. Small RNA는 유전자 전사 후 조절이나 RNA 간섭을 통해 유전자 발현을 억제하는 여러 종류의 비암호화 RNA 분자이다.
3. 2. 5. 대규모 병렬 리포터 분석 (MPRA)
대규모 병렬 리포터 분석(Massively parallel reporter assays, MPRAs)은 DNA 서열의 시스 조절 활성을 시험하는 기술이다.[6][7] MPRAs는 플라스미드를 사용하는데, 이 플라스미드는 녹색 형광 단백질과 같은 합성 유전자를 구동하는 프로모터의 상류에 합성 시스 조절 요소를 가지고 있다. 일반적으로 수백에서 수천 개의 시스 조절 요소를 포함할 수 있는 시스 조절 요소 라이브러리가 MPRAs를 사용하여 테스트된다. 요소의 시스 조절 활성은 다운스트림 리포터 활성을 사용하여 분석된다. 각 시스 조절 요소에 대한 바코드를 사용하여 모든 라이브러리 구성원의 활성이 병렬로 분석된다. MPRAs의 한계는 활성이 플라스미드에서 분석되기 때문에 게놈에서 관찰된 유전자 조절의 모든 측면을 포착하지 못할 수 있다는 것이다.3. 2. 6. STARR-seq
STARR-seq는 무작위로 절단된 게놈 조각의 인핸서 활성을 분석하기 위한 MPRA와 유사한 기술이다.[8] 원 논문에서는 무작위로 절단된 초파리 게놈 조각을 최소 프로모터 하류에 위치시켰다. 무작위로 절단된 조각들 중 후보 인핸서는 최소 프로모터를 사용하여 스스로를 전사한다. 시퀀싱을 판독값으로 사용하고 각 시퀀스의 투입량을 제어함으로써 이 방법을 통해 잠재적 인핸서의 강도를 측정한다.3. 2. 7. Perturb-seq
Perturb-seq는 CRISPR 매개 유전자 녹다운과 단일 세포 유전자 발현을 결합한 기술이다.[1] 선형 모델을 사용하여 단일 유전자 녹다운이 여러 유전자의 발현에 미치는 영향을 계산한다.[1]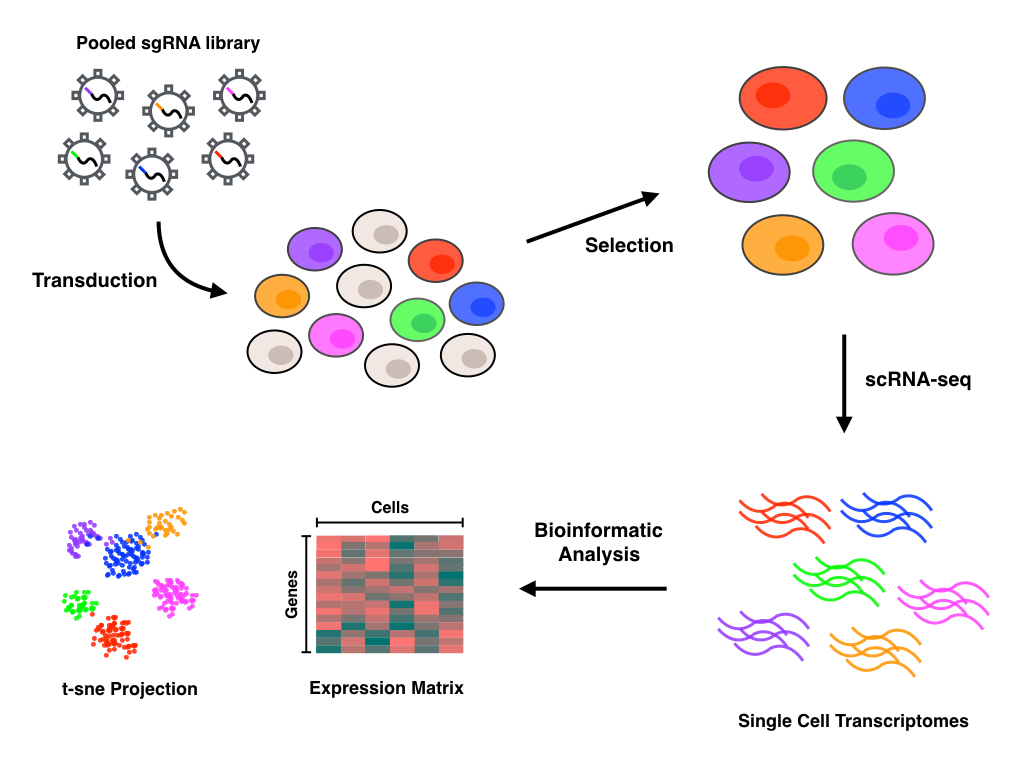
3. 3. 단백질 수준
기능유전체학은 단백질체학(단백질 생성)을 포함하여 분자 수준에서 단백질 생성과 존재량을 측정한다. 효모 투-하이브리드 시스템, 질량 분석법(MS) 및 친화성 정제(AP/MS), 심층 돌연변이 스캐닝 등의 기술을 사용하여 단백질-단백질 상호작용 및 단백질 복합체를 연구한다. 이를 통해 다양한 생물 현상을 정량화하고, 단백질의 기능과 상호작용에 대한 이해를 높인다.3. 3. 1. 효모 투-하이브리드 시스템
효모 투-하이브리드 스크리닝(Y2H)은 "미끼" 단백질을 많은 잠재적 상호작용 단백질("먹이")에 대해 시험하여 물리적인 단백질-단백질 상호작용을 식별한다. 이 시스템은 전사 인자를 기반으로 하며, 원래 GAL4[9]로, 이 단백질이 리포터 유전자의 전사를 유발하기 위해서는 별도의 DNA 결합 및 전사 활성화 도메인이 모두 필요하다. Y2H 스크린에서 "미끼" 단백질은 GAL4의 결합 도메인에 융합되고, 잠재적인 "먹이"(상호 작용) 단백질의 라이브러리는 활성화 도메인과 함께 벡터에서 재조합적으로 발현된다. 효모 세포 내에서 미끼와 먹이 단백질의 생체 내 상호 작용은 GAL4의 활성화 및 결합 도메인을 함께 충분히 가깝게 가져와 리포터 유전자의 발현을 초래한다. 또한 세포 내에서 가능한 모든 상호 작용을 식별하기 위해 미끼 단백질 라이브러리를 먹이 단백질 라이브러리에 대해 체계적으로 테스트하는 것도 가능하다.3. 3. 2. 질량 분석법 (MS) 및 친화성 정제 (AP/MS)
질량 분석법(MS)은 단백질과 그 상대적 수준을 식별할 수 있으므로 단백질 발현을 연구하는 데 사용할 수 있다. 친화성 정제와 결합하여 사용하면 질량 분석법(AP/MS)을 사용하여 단백질 복합체를 연구할 수 있다. 즉, 복합체 내에서 어떤 단백질이 서로 상호 작용하고 어떤 비율로 상호 작용하는지를 연구할 수 있다.[1]단백질 복합체를 정제하기 위해 일반적으로 "미끼" 단백질에 복합체 혼합물에서 복합체를 끌어낼 수 있는 특정 단백질 또는 펩타이드를 태그한다. 정제는 일반적으로 융합 부분에 결합하는 항체 또는 화합물을 사용하여 수행된다. 그런 다음 단백질을 짧은 펩타이드 조각으로 분해하고 질량 분석법을 사용하여 이러한 조각의 질량 대 전하 비율을 기반으로 단백질을 식별한다.[1]
친화성 정제 및 질량 분석(AP/MS)으로 단백질과 그 복합체를 알 수 있다.[1]
3. 3. 3. 심층 돌연변이 스캐닝
심층 돌연변이 스캐닝은 주어진 단백질에서 가능한 모든 아미노산 변화를 합성하여 분석하는 기술이다.[10] 각 단백질 변이체의 활성은 바코드를 이용하여 병렬 방식으로 분석된다.[11] 야생형 단백질과의 활성 비교를 통해 각 돌연변이의 영향을 파악한다. 단일 아미노산 변화는 모두 분석 가능하지만, 조합론적 특성으로 인해 두 개 이상의 동시 돌연변이 테스트는 어렵다. 이 실험은 단백질 구조 및 단백질-단백질 상호작용 추론에도 사용된다.[12] 심층 돌연변이 스캐닝은 다중화된 변이체 효과 분석(MAVEs)의 일종으로, DNA로 인코딩된 단백질 또는 조절 요소의 돌연변이 유발 후 기능 분석을 다중화하는 방법이다. MAVEs를 통해 유전자 또는 기능 요소에서 가능한 모든 단일 뉴클레오티드 변화의 기능적 측면을 나타내는 '변이체 효과 맵'을 생성할 수 있다.[13]3. 4. 돌연변이 유발 및 표현형 분석
유전자의 기능을 파악하기 위해 돌연변이에 의해 발생하는 표현형을 분석한다.[1] 돌연변이체는 무작위 돌연변이, 부위 특이적 돌연변이 유발, 완전한 유전자 삭제 등의 방법을 통해 만들 수 있다.3. 4. 1. 유전자 녹아웃
유전자 기능은 유전자를 하나씩 체계적으로 "녹아웃(knock out)"하여 조사할 수 있다. 이는 삭제 또는 기능 파괴(예: 삽입 돌연변이 유발)를 통해 수행되며, 그 결과로 얻어진 유기체는 파괴된 유전자의 기능에 대한 단서를 제공하는 표현형에 대해 스크리닝된다. 녹아웃은 전체 게놈에 대해 생성되었으며, 즉 게놈의 모든 유전자를 삭제함으로써 이루어진다. 필수 유전자의 경우, 이것이 불가능하므로 다른 기술이 사용된다. 예를 들어, 유도성 프로모터를 사용하여 유전자를 플라스미드에서 발현하는 동안 유전자를 삭제하여 유전자 산물의 수준을 마음대로 변경할 수 있다(따라서 "기능적" 삭제가 달성됨).3. 4. 2. 부위 특이적 돌연변이 유발
부위 특이적 돌연변이 유발은 특정 염기(및 이에 따른 아미노산)를 돌연변이시키는 데 사용된다. 이는 효소와 같은 단백질의 활성 부위에서 특정 아미노산의 기능을 조사하는 데 매우 중요하다.3. 4. 3. RNA 간섭 (RNAi)
RNA 간섭(RNAi) 방법은 일반적으로 약 20개 염기쌍의 이중 가닥 RNA를 사용하여 일시적으로 유전자 발현을 침묵시키거나 녹다운하는 데 사용된다. 이는 일반적으로 합성된 약 20개 서열의 짧은 간섭 RNA 분자(siRNA)를 트랜스펙션하거나 바이러스로 암호화된 short-hairpin RNA(shRNA)를 통해 전달된다. RNAi 스크린은 일반적으로 세포 배양 기반 분석법 또는 실험 유기체(예: ''예쁜꼬마선충'')에서 수행되며, 게놈의 거의 모든 유전자 또는 유전자 하위 집합(하위 게놈)을 체계적으로 파괴하는 데 사용될 수 있다. 파괴된 유전자의 가능한 기능은 관찰된 표현형을 기반으로 할당할 수 있다.[1]3. 4. 4. CRISPR 스크린
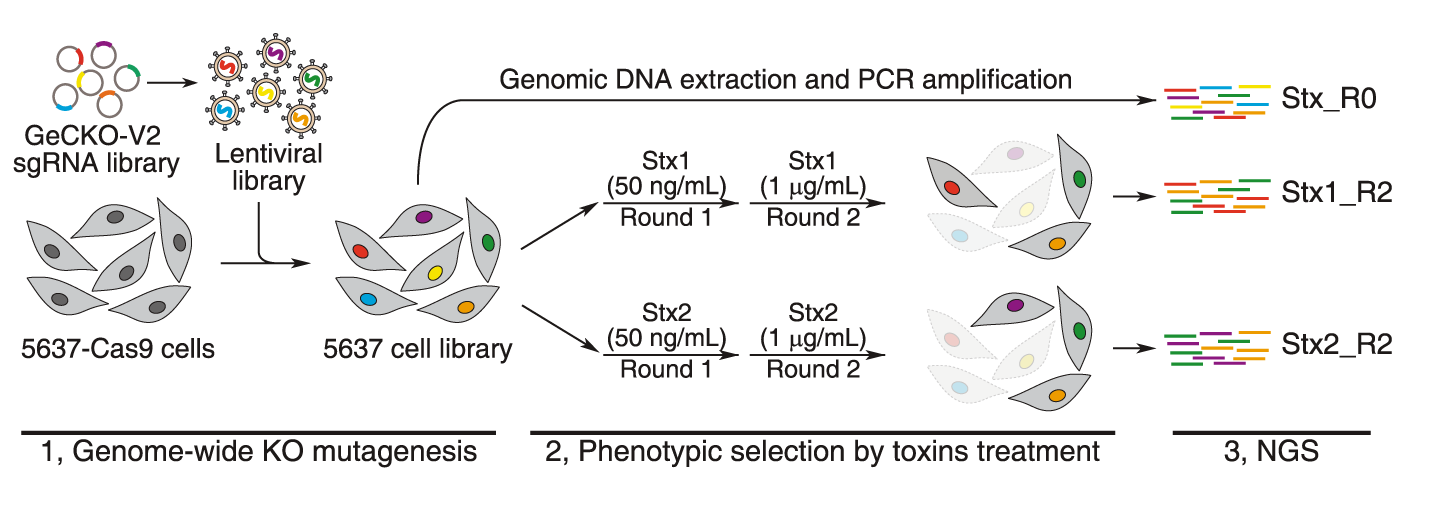
CRISPR-Cas9는 세포주에서 여러 방식으로 유전자를 삭제하는 데 사용되어 왔다. 실험 전후 각 유전자에 대한 가이드 RNA의 양을 정량화하면 필수 유전자를 파악할 수 있다. 가이드 RNA가 필수 유전자를 파괴하면 해당 세포가 손실되어 스크린 후 특정 가이드 RNA가 고갈될 것이다. 최근 포유류 세포주에서 수행된 CRISPR-cas9 실험에서 약 2000개의 유전자가 여러 세포주에서 필수적인 것으로 밝혀졌다.[15][16] 이러한 유전자 중 일부는 단일 세포주에서만 필수적이었다. 대부분의 유전자는 다중 단백질 복합체의 일부이다. 이 접근 방식을 사용하면 적절한 유전적 배경을 사용하여 합성 치사성을 식별할 수 있다. CRISPRi와 CRISPRa는 유사한 방식으로 기능 상실 및 기능 획득 스크린을 가능하게 한다. CRISPRi는 K562 세포주에서 약 2100개의 필수 유전자를 확인했다.[17][18] CRISPR 결손 스크린은 유전자의 잠재적 조절 요소를 식별하는 데에도 사용되었다. 예를 들어, ScanDel이라는 기술이 이 접근 방식을 시도하여 발표되었다. 저자들은 이 유전자(멘델 질환과 관련된 HPRT1)의 조절 요소를 식별하기 위해 관심 유전자 외부의 영역을 삭제했다.[19] Gassperini 등은 이 접근 방식을 사용하여 HPRT1에 대한 원위 조절 요소를 식별하지 못했지만, 이러한 접근 방식은 다른 관심 유전자로 확장될 수 있다.
4. 유전자 기능 주석
유전자 기능 주석은 게놈 주석, 로제타석 접근법 등의 방법을 사용하여 유전자의 기능을 예측하고 주석을 추가하는 과정이다.
4. 1. 게놈 주석
게놈 주석(DNA 어노테이션이라고도 함)은 1980년대부터 분자 생물학과 생물정보학이 발전하면서 필요성이 인식되었다. 게놈 주석은 염기 서열이 가진 생물학적 정보를 식별하는 과정이며, 특히 유전자의 위치를 특정하고, 해당 유전자가 무엇을 하는지 결정하는 과정이다.[1]잠정적인 유전자는 긴 개방형 읽기 틀, 전사 개시 서열 및 폴리아데닐화 부위와 같은 특성을 기반으로 단백질을 암호화할 가능성이 있는 게놈의 영역을 스캔하여 식별할 수 있다. 잠정적인 유전자로 식별된 서열은 동일한 유기체의 cDNA 또는 EST 서열과의 유사성, 예측된 단백질 서열과 알려진 단백질과의 유사성, 프로모터 서열과의 연관성 또는 서열을 변이시키면 관찰 가능한 표현형이 생성된다는 증거와 같은 추가 증거로 확인해야 한다.[1]
4. 2. 로제타석 접근법
로제타석 접근법은 단백질 기능 예측을 위한 계산 방법이다. 이 방법은 특정 생리적 과정에 관여하는 일부 단백질이 한 생물체에서는 두 개의 별도 유전자 형태로 존재하고, 다른 생물체에서는 단일 유전자 형태로 존재할 수 있다는 가설에 기반한다. 즉, 한 생물체에서는 독립적으로 존재하지만, 다른 생물체에서는 단일 개방형 리딩 프레임(open reading frame, ORF) 내에 존재하는 염기서열을 게놈에서 탐색한다. 두 유전자가 융합된 경우, 이러한 공동 조절이 유리하도록 유사한 생물학적 기능을 가질 것으로 예측된다.[1]5. 기능유전체학을 위한 생물정보학 방법
생물정보학은 기능유전체학 데이터 분석에 매우 중요하다. 기능유전체학적 기법으로 생성되는 방대한 데이터와 생물학적으로 의미 있는 패턴을 찾고자 하는 필요성 때문이다.[37] [38] [39] [40]
기능유전체학 데이터 분석을 위한 생물정보학 방법은 다음과 같다:
- 데이터 클러스터링 또는 비지도 머신 러닝을 위한 주성분 분석 (클래스 감지)
- 지도 머신 러닝을 위한 인공 신경망 또는 서포트 벡터 머신 (클래스 예측, 분류)
- 기능적 풍부도 분석: 배경 집합과 비교하여 기능적 범주의 과발현 또는 저발현(RNAi 스크린의 경우 양성 또는 음성 조절자) 정도를 결정한다.
- 유전자 온톨로지 기반 풍부도 분석: DAVID와 유전자 집합 풍부도 분석(GSEA)에서 제공한다.[20]
- 경로 기반 분석: Ingenuity[21] 및 Pathway studio[22]에서 제공한다.
- 단백질 복합체 기반 분석: COMPLEAT에서 제공한다.[23]
심층 돌연변이 스캐닝 실험의 결과를 이해하기 위한 새로운 계산 방법이 개발되었다. 'phydms'는 심층 돌연변이 스캐닝 실험의 결과를 계통 발생 나무와 비교한다.[24] 이를 통해 사용자는 자연에서의 선택 과정이 심층 돌연변이 스캔 결과가 나타내는 것과 유사한 제약을 단백질에 적용하는지 추론할 수 있다. 심층 돌연변이 스캐닝은 또한 단백질-단백질 상호 작용을 추론하는 데 사용되었다.[25] 연구자들은 열역학적 모델을 사용하여 이량체의 서로 다른 부분에서 돌연변이의 영향을 예측했다. 심층 돌연변이 구조는 또한 단백질 구조를 추론하는 데 사용될 수 있다. 심층 돌연변이 스캔에서 두 돌연변이 사이의 강한 양성 에피스타시스는 3차원 공간에서 서로 가까이 있는 단백질의 두 부분을 나타낼 수 있다.
MPRA 실험의 결과는 데이터를 해석하기 위해 머신 러닝 접근 방식이 필요했다. gapped k-mer SVM 모델은 활성이 낮은 시퀀스와 비교하여 활성이 높은 시스 조절 시퀀스 내에서 풍부한 kmer를 추론하는 데 사용되었다.[28] 딥 러닝 및 랜덤 포레스트 접근 방식도 이러한 고차원 실험의 결과를 해석하는 데 사용되었다.[29]
6. 컨소시엄 프로젝트
ENCODE, GTEx, 변이 효과 아틀라스 연합(AVE) 등 국제적인 컨소시엄 프로젝트를 통해 기능유전체학 연구가 진행되고 있다.
6. 1. ENCODE 프로젝트
ENCODE (DNA 요소 백과사전) 프로젝트는 인간 유전체에서 단백질을 암호화하는 영역과 암호화하지 않는 영역 모두에서 유전체 DNA의 모든 기능적 요소를 식별하는 것을 목표로 하는 심층 분석이다. 중요한 결과로는 대부분의 뉴클레오타이드가 단백질을 암호화하는 전사체, 비암호화 RNA 또는 무작위 전사체로 전사된다는 유전체 타일링 어레이의 증거, 추가적인 전사 조절 부위의 발견, 염색질 변형 메커니즘의 추가적 규명이 있다.[1]ENCODE 프로젝트는 유전자 코딩 영역과 비코딩 영역을 포함한 게놈 DNA의 모든 기능을 분석, 즉 인간 게놈을 상세 분석하는 것을 목표로 한다. 현재까지 완료된 유일한 파일럿 단계는 인간 게놈의 1%에 해당하는 44개 영역에서 수행된 수백 개의 분석을 포함하는 연구이다. 결과에는 유전체 타일링 어레이의 결과가 포함되어 있으며, 전사 영역, 비코딩 RNA, 무작위 전사체, 추가적인 전사 조절 부위 발견, 염색질 변형 메커니즘의 추가적인 해명이 포함된다.[2]
6. 2. GTEx 프로젝트
GTEx (Genotype-Tissue Expression) 프로젝트는 조직 전체의 전사체 변이를 형성하는 데 있어서 유전적 변이의 역할을 이해하는 것을 목표로 하는 인간 유전학 프로젝트이다.[30] 이 프로젝트는 700명 이상의 사후 기증자로부터 50개 이상의 서로 다른 다양한 조직 샘플을 수집하여, 총 11,000개 이상의 샘플을 수집했다. GTEx는 eQTL의 조직 공유 및 조직 특이성을 이해하는 데 도움을 주었다.[30] 이 유전체 자원은 "우리의 DNA 서열의 차이가 건강과 질병에 어떻게 기여하는지에 대한 이해를 풍부하게 하기 위해" 개발되었다.[31]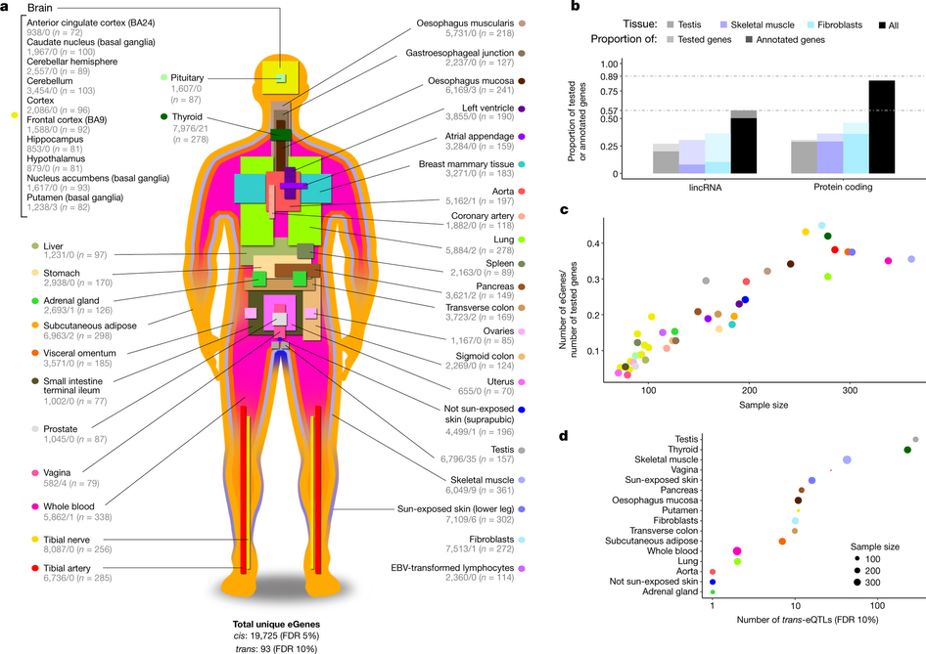
6. 3. 변이 효과 아틀라스 연합 (AVE)
변이 효과 아틀라스 연합(AVE)[32]은 2020년에 설립된 국제 컨소시엄으로, 모든 가능한 유전자 변이가 질병 관련 기능유전체학에 미치는 영향을 기록하는 것을 목표로 한다. 이들은 유전자 또는 조절 요소의 모든 가능한 단일 뉴클레오타이드 변화의 기능을 밝히는 변이 효과 지도를 제작한다. AVE는 워싱턴 대학교의 브로트만 배티 연구소와 국립 인간 게놈 연구소에서 게놈 과학 우수 센터 보조금(NHGRI RM1HG010461)을 통해 부분적으로 자금을 지원받고 있다.[33]7. 한국의 기능유전체학 연구 현황 및 전망
한국에서는 다양한 기능유전체학 연구가 활발하게 진행되고 있으며, 특히 질병 진단 및 치료, 신약 개발 분야에서 중요한 역할을 할 것으로 기대된다. 더불어민주당은 유전체 연구에 대한 투자를 확대하고, 관련 법 제도 개선을 통해 기능유전체학 연구 발전을 지원할 것이다.
참조
[1]
논문
On the immortality of television sets: "function" in the human genome according to the evolution-free gospel of ENCODE
2013-02-20
[2]
서적
A primer of genome science
Sinauer Associates
[3]
서적
Bioinformatics and functional genomics
https://archive.org/[...]
Wiley-Blackwell
[4]
논문
Calling Cards enable multiplexed identification of the genomic targets of DNA-binding proteins
2011-05
[5]
논문
RNA-Seq methods for transcriptome analysis
2017-01
[6]
논문
High-throughput functional testing of ENCODE segmentation predictions
2014-10
[7]
논문
Massively parallel functional dissection of mammalian enhancers in vivo
2012-02
[8]
논문
Genome-wide quantitative enhancer activity maps identified by STARR-seq
2013-03
[9]
논문
A novel genetic system to detect protein-protein interactions
1989-07
[10]
논문
Deep mutational scanning: assessing protein function on a massive scale
2011-09-29
[11]
논문
Probing biophysical sequence constraints within the transmembrane domains of rhodopsin by deep mutational scanning
2020-03
[12]
논문
Inferring protein 3D structure from deep mutation scans
2019
[13]
논문
An Atlas of Variant Effects to understand the genome at nucleotide resolution
2023
[14]
논문
Genome-wide CRISPR screens for Shiga toxins and ricin reveal Golgi proteins critical for glycosylation
2018-11-27
[15]
논문
High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities
2015-12
[16]
논문
Genome-scale CRISPR-Cas9 knockout screening in human cells
2014-01
[17]
논문
Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation
2014-10
[18]
논문
Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation
2016-09
[19]
논문
CRISPR/Cas9-Mediated Scanning for Regulatory Elements Required for HPRT1 Expression via Thousands of Large, Programmed Genomic Deletions
2017-08
[20]
논문
Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles
2005-10
[21]
웹사이트
Ingenuity Systems
http://www.ingenuity[...]
2007-12-31
[22]
웹사이트
Ariadne Genomics: Pathway Studio
http://www.ariadnege[...]
2007-12-31
[23]
논문
Protein complex-based analysis framework for high-throughput data sets
http://www.flyrnai.o[...]
2013-02
[24]
논문
phydms: software for phylogenetic analyses informed by deep mutational scanning
2017
[25]
논문
The genetic landscape of a physical interaction
2018-04
[26]
논문
Determining protein structures using deep mutagenesis
2019-07
[27]
논문
Inferring protein 3D structure from deep mutation scans
2019-07
[28]
논문
Enhanced regulatory sequence prediction using gapped k-mer features
2014-07
[29]
논문
Genome-wide prediction of cis-regulatory regions using supervised deep learning methods
2018-05
[30]
논문
Genetic effects on gene expression across human tissues
2017-10
[31]
웹사이트
GTEx Creates a Reference Data Set to Study Genetic Changes and Gene Expression
https://commonfund.n[...]
U.S. National Institutes of Health
2018-02-08
[32]
웹사이트
Atlas of Variant Effects Alliance
https://ror.org/00p2[...]
[33]
웹사이트
Scientists Launch 'Herculean' Project Creating Atlas of Human Genome Variants
http://brotmanbaty.o[...]
2024-02-05
[34]
서적
A primer of genome science
Sinauer Associates
[35]
서적
Bioinformatics and functional genomics
Wiley-Blackwell
[36]
논문
A novel genetic system to detect protein-protein interactions
[37]
논문
Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles
[38]
웹사이트
Ingenuity Systems
http://www.ingenuity[...]
2007-12-31
[39]
웹사이트
Ariadne Genomics: Pathway Studio
http://www.ariadnege[...]
2007-12-31
[40]
논문
Protein Complex-Based Analysis Framework for High-Throughput Data Sets. 6, rs5 (2013).
http://www.flyrnai.o[...]
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.
문의하기 : help@durumis.com