HIV-단백질분해효소 억제제의 발견과 발전
"오늘의AI위키"의 AI를 통해 더욱 풍부하고 폭넓은 지식 경험을 누리세요.
1. 개요
HIV 단백질분해효소 억제제는 HIV 감염 치료를 위해 개발된 약물로, HIV의 생존에 필수적인 단백질분해효소의 작용을 억제하여 바이러스 복제를 막는다. 1980년대 HIV 발견 이후, 사퀴나비르를 시작으로 리토나비르, 인디나비르 등 다양한 억제제가 개발되었으며, 약물 내성을 극복하기 위한 연구가 지속되고 있다. 이러한 억제제는 HIV의 생활사 중 바이러스 단백질의 분해를 방해하여 바이러스 입자가 성숙하는 것을 막는 방식으로 작용한다. 최근에는 약물 내성을 줄이고 치료 효과를 높이기 위해 새로운 제형 개발 및 기존 약물과의 병용 요법 연구가 진행되고 있다.
더 읽어볼만한 페이지
- HIV/AIDS - 아바카비르/라미부딘/지도부딘
HIV 감염 치료에 사용되는 복합 약물인 아바카비르/라미부딘/지도부딘은 과민성 반응, 젖산 산증, 간 비대증, 지질 이상, 골수 억제, 심근경색 위험 증가, 면역재구성염증증후군(IRIS), 빈혈, 호중구 감소증, 지방위축증과 같은 다양한 부작용을 유발할 수 있다. - HIV/AIDS - 래리 크레이머
래리 크레이머는 미국의 극작가, 소설가, 에이즈 활동가로, 영화 각본가로 활동하며 에이즈 위기에 대응하여 단체를 설립하고 작품을 통해 에이즈 문제에 대한 사회적 인식을 높이는 데 기여했으며 2020년에 사망했다.
| HIV-단백질분해효소 억제제의 발견과 발전 | |
|---|---|
| 약물 정보 | |
| ATC 코드 | J05AE01 (사퀴나비르) J05AE02 (리토나비르) J05AE03 (인데나비르) J05AE04 (넬피나비르) J05AE05 (암프레나비르) J05AE07 (로피나비르) J05AE08 (아타자나비르) J05AE09 (티프라나비르) J05AE10 (다루나비르) J05AE11 (포삼프레나비르) J05AE12 (사퀴나비르/리토나비르) J05AE13 (아타자나비르/리토나비르) J05AE14 (다루나비르/코비시스타트) J05AE15 (로피나비르/리토나비르) J05AE16 (넬피나비르/리토나비르) J05AE17 (팁라나비르/리토나비르) J05AE18 (브레카나비르) J05AE19 (베클레부비르) J05AE51 (리토나비르, 병용) |
| 단백질 표적 | HIV-1 프로테아제 |
| 투여 경로 | 경구 |
| 작용 기전 | |
| 작용 방식 | HIV-1 프로테아제 억제 |
| 상세 설명 | HIV-1 프로테아제는 바이러스 복제에 필수적인 단백질로, 이를 억제하여 바이러스의 성장을 막음. |
| 관련 질병 | |
| 질병 | HIV 감염 |
| 연구 | |
| 연구 분야 | 화학 생물학 약물 설계 |
| 참고 문헌 | |
| 참고 문헌 목록 | "Cuccioloni M, Mozzicafreddo M, Bonfili L, Cecarini V, Eleuteri AM, Angeletti M (2009). “Natural occurring polyphenols as template for drug design. Focus on serine proteases”. Chemical Biology & Drug Design. 74 (1): 1–15. doi:10.1111/j.1747-0285.2009.00836.x. PMID 19519739. S2CID 21193839.: 폴리페놀을 기반으로 한 약물 설계 연구" "Chen X, Kempf DJ, Li L, Sham HL, Vasavanonda S, Wideburg NE, Saldivar A, Marsh KC, McDonald E, Norbeck DW (2003). “Synthesis and SAR studies of potent HIV protease inhibitors containing novel dimethylphenoxyl acetates as P2 ligands”. Bioorganic & Medicinal Chemistry Letters. 13 (21): 3657–60. doi:10.1016/j.bmcl.2003.08.043. PMID 14552751.: 새로운 디메틸페녹시 아세테이트 P2 리간드를 포함하는 강력한 HIV 프로테아제 억제제 합성 및 SAR 연구" "Adachi M, Ohhara T, Kurihara K, Tamada T, Honjo E, Okazaki N, Arai S, Shoyama Y, Kimura K, Matsumura H, Sugiyama S, Adachi H, Takano K, Mori Y, Hidaka K, Kimura T, Hayashi Y, Kiso Y, Kuroki R (2009). “Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography”. Proceedings of the National Academy of Sciences. 106 (12): 4641–6. Bibcode:2009PNAS..106.4641A. doi:10.1073/pnas.0809400106. PMC 2660780. PMID 19273847.: 고해상도 X선 및 중성자 결정학으로 결정된 강력한 억제제 KNI-272와 복합체를 이룬 HIV-1 프로테아제의 구조" "Yanchunas J Jr, Langley DR, Tao L, Rose RE, Friborg J, Colonno RJ, Doyle ML (2005). “Molecular basis for increased susceptibility of isolates with atazanavir resistance-conferring substitution I50L to other protease inhibitors”. Antimicrobial Agents and Chemotherapy. 49 (9): 3825–32. doi:10.1128/AAC.49.9.3825-3832.2005. PMC 1195399. PMID 16127059.: 아타자나비르 내성 치환 I50L을 갖는 분리주의 다른 프로테아제 억제제에 대한 감수성 증가의 분자적 기초" "Brower ET, Bacha UM, Kawasaki Y, Freire E (2008). “Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use”. Chemical Biology & Drug Design. 71 (4): 298–305. doi:10.1111/j.1747-0285.2008.00647.x. PMID 18312292. S2CID 8461472.: 임상에서 사용되는 HIV-1 프로테아제 억제제에 의한 HIV-2 프로테아제 억제" |
2. 역사
HIV 감염은 1981년 처음 보고되었으며, 1985년에 후천성면역결핍증후군(AIDS)의 원인으로 밝혀지고 전체 게놈이 분석되었다.[47][48][7][6] 이러한 발견은 HIV 치료를 위한 선택적 효소 억제제 개발의 기반이 되었다.[48][6] HIV 프로테아제가 발견된 후, 1987년에 첫 수용체 대항제가 보고되었고, 약 10년 뒤인 1995년에는 최초의 HIV 프로테아제 억제제인 사퀴나비르가 처방약으로 승인받았다.[51][46][10][6] 이후 리토나비르와 인디나비르 등 다른 억제제들이 연이어 승인되었다.[46][6]
2. 1. HIV의 발견과 AIDS의 확산
HIV는 레트로바이러스과에 속하는 렌티바이러스이며, 크게 HIV-1과 HIV-2 두 가지 주요 종으로 나뉜다. 이 중 HIV-1이 전 세계적인 유행병의 대부분을 일으키는 반면, HIV-2는 주로 서아프리카 지역에 분포하는 경향을 보인다.[6] HIV 감염 사례는 1981년 미국 샌프란시스코와 뉴욕에서 처음으로 보고되었다.[7]1985년에 이르러 HIV가 후천성면역결핍증후군(AIDS)을 일으키는 원인 바이러스임이 공식적으로 확인되었고, 곧이어 바이러스의 전체 게놈 서열이 밝혀졌다. 이러한 지식은 HIV 감염 치료를 위한 선택적인 효소 억제제 개발의 길을 열었다.[6]
HIV-1과 비교했을 때, HIV-2는 감염될 확률이 다소 낮고, 감염 후 AIDS로 병이 진행되는 속도도 더 느린 특징을 가지고 있다.[7] 일반적으로 'HIV'라고 할 때는 대부분 HIV-1을 가리킨다.[8]
2. 2. HIV 프로테아제 억제제의 개발
HIV-1 프로테아제는 가장 잘 알려진 아스파르트산 프로테아제 중 하나이며, AIDS 치료를 위한 중요한 표적으로 여겨진다.[50][9] HIV 프로테아제가 발견된 이후, 첫 번째 효소 억제제가 시장에 출시되기까지는 단 10년이 걸렸다.[51][10]1987년, HIV 프로테아제에 대한 매우 선택적인 수용체 대항제가 처음으로 보고되었다.[46][6] 사퀴나비르(saquinavir)는 1989년에 1상 임상 시험을 시작했으며, 1995년 미국 식품의약국(FDA)의 승인을 받아 최초의 HIV 프로테아제 억제제 처방약이 되었다.[46][6] 그로부터 4개월 후, 리토나비르(ritonavir)와 인디나비르(indinavir)라는 두 가지 다른 프로테아제 억제제도 승인을 받았다.[46][6]
2009년까지 HIV 치료를 위해 총 10종의 프로테아제 억제제가 시장에 출시되었다.[46][6] 그러나 이 중 암프레나비르(amprenavir)는 2004년에 시장에서 철수되었다.[46][52][11]
3. HIV의 생활사
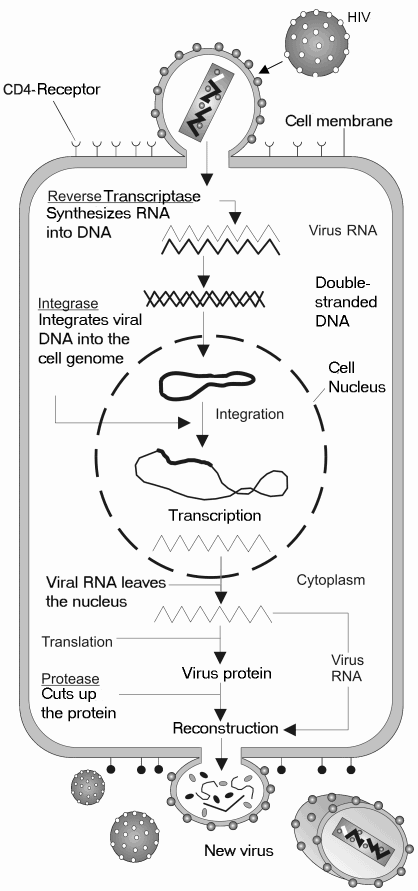
HIV는 유전물질로 RNA를 사용하는 레트로바이러스의 일종으로, 주로 표면에 CD4라는 항원을 가진 T 세포를 감염시킨다. HIV가 세포를 감염시키기 위해서는 바이러스와 표적 세포의 세포막이 서로 융합되어야 한다.[53][12]
감염 과정은 다음과 같다.
# 부착 및 결합: 바이러스의 외피 단백질(gp120, gp41)이 먼저 표적 세포 표면의 CD4 수용체와 결합한다. 이후 바이러스는 CXCR4나 CCR5와 같은 케모카인 공수용체(coreceptor)에 추가로 결합한다. 이 결합은 바이러스 외피 단백질의 구조적 변화를 유발한다.
# 융합 및 침투: 구조 변화를 통해 바이러스 외피와 세포막이 융합되고, 이를 통해 바이러스의 핵심 부분인 캡시드가 세포 안으로 들어갈 수 있는 통로가 만들어진다.[54][13]
# 역전사: 세포 안으로 들어온 바이러스의 RNA는 바이러스 자체 효소인 역전사 효소에 의해 DNA 형태로 변환된다.
# 통합: 역전사된 바이러스 DNA는 세포의 핵 안으로 이동하여, 또 다른 바이러스 효소인 통합효소(integrase)에 의해 숙주 세포의 유전체(DNA)에 끼어 들어간다.
# 복제: 숙주 세포가 활성화되어 자신의 DNA를 복제하고 단백질을 만들 때, 삽입된 바이러스 DNA도 함께 mRNA로 전사된다.
# 조립: 전사된 mRNA는 바이러스 단백질로 번역된다. 이때, 바이러스 효소인 HIV 단백질 분해 효소(protease)가 생성된 긴 단백질 가닥(전구체 단백질)을 잘라 성숙한 바이러스 구성 단백질 조각들을 만든다. 이 단백질 조각들과 바이러스 RNA는 세포 표면 근처에서 조립되어 새로운 비리온(virion, 바이러스 입자)을 형성한다.
# 방출: 새로 만들어진 비리온은 세포막을 이용하여 출아 방식으로 세포 밖으로 빠져나와 다른 세포들을 감염시킨다.
이 과정에서 숙주 세포는 유전 물질이 변형되고, 새로운 바이러스 입자가 빠져나가면서 세포막 등이 손상되어 결국 파괴된다.[55][12]
4. HIV 프로테아제 억제제의 작용 기전
HIV의 복제 과정에는 여러 단계가 있으며, 이 중 일부를 방해하여 바이러스 증식을 막을 수 있다. 특히 중요한 단계는 HIV 프로테아제가 바이러스의 폴리펩타이드 전구체를 절단하여 성숙한 효소와 구조 단백질을 만드는 과정이다.[53] HIV 프로테아제 억제제는 바이러스의 아스파르트산 프로테아제 활성 부위에 결합하여 그 작용을 경쟁적으로 억제하는 펩타이드 유사 물질이다. 이 약물들은 바이러스의 핵심적인 구조 및 효소 성분을 포함하는 HIV Gag-Pol 폴리펩타이드의 분해를 막는다. 결과적으로, 새로 생성되는 HIV 입자는 감염력을 갖지 못하는 미성숙한 상태로 남게 된다.[46]
한편, 프로테아제 억제제는 지방 세포의 대사를 변화시켜 지방 이상증과 같은 부작용을 유발할 수 있다. 이러한 부작용의 정확한 발생 기전은 아직 연구 중이지만, 몇 가지 가설이 제시되었다. 예를 들어, 프로테아제 억제제가 지방세포의 분화와 중성지방 축적을 억제하고, 지방 분해를 증가시킬 수 있다는 설명이 있다. 또한, 인슐린에 의해 조절되는 포도당 흡수에 미치는 영향도 지방 이상증과 관련될 수 있다. 한 이론에 따르면, 프로테아제 억제제는 인슐린 신호 전달의 초기 단계를 방해하여, 인슐린 자극에 따른 IRS-1 단백질의 티로신 인산화를 감소시킬 수 있다. 더불어, HIV 프로테아제 억제제가 아디포넥틴의 분비를 줄이고 인터루킨-6의 발현을 유도하여, 결과적으로 인슐린에 의한 포도당 흡수를 억제할 수 있다는 가능성도 제기된다.[56]
5. HIV 프로테아제 억제제의 설계 및 발전
HIV 단백질 분해효소 억제제는 효소의 실제 기질이 전이 상태에 있을 때의 구조를 모방하도록 설계되었다.[57][15] 핵심 원리는 효소가 절단하는 펩타이드 결합(–NH-CO–) 대신, 효소가 절단할 수 없는 하이드록시에틸렌 그룹(-CH2-CH(OH)-)이나 이와 유사한 구조로 대체하는 것이다.[57] HIV 아스파르트산 단백질 분해효소의 활성 부위에 잘 맞도록 만들어졌으며, 이 효소의 작용 기전에 대한 지식을 바탕으로 합리적으로 설계되었다.[15] 여러 구조 중 하이드록시에틸아민(hydroxyethylamine)은 전이 상태를 효과적으로 모방하는 것으로 밝혀졌고, 이는 최초의 단백질 분해효소 억제제인 사퀴나비르(saquinavir)의 발견으로 이어졌다.[57][15] 사퀴나비르는 1995년에 출시되었다.[60]
사퀴나비르 이후, 다양한 설계 전략을 통해 여러 HIV 단백질 분해효소 억제제가 개발되었다. 리토나비르(ritonavir, 1996년 출시)는 효소 결합 부위의 C2 대칭성에 맞게 설계되었고,[46][60] 인디나비르(indinavir, 1996년 출시)는 분자 모델링과 X선 결정 구조 분석을 통해 설계되었다.[61][60] 넬피나비르(nelfinavir, 1997년 출시)는 펩티도모방체가 아닌 최초의 억제제로, 비펩티드 치환기를 도입하여 개발되었다.[61][60] 암프레나비르(amprenavir, 1999년 출시)는 구조를 단순화하고 수용성을 개선하여 경구 생체 이용률을 높였다.[61][60] 로피나비르(lopinavir, 2000년 출시)는 약물 내성 바이러스에서 나타나는 돌연변이에 대응하기 위해 설계되었다.[61][60] 포삼프레나비르(fosamprenavir, 2003년 출시)는 암프레나비르의 전구약물로 개발되어 용해도와 생체 이용률을 개선했다.[64][60] 아타자나비르(atazanavir, 2003년 출시)는 아자펩타이드 구조를 기반으로 설계되었으며,[60] 티프라나비르(tipranavir, 2005년 출시)는 다른 억제제와 달리 비펩티드 쿠마린 구조에서 유래되었다.[66][60] 다루나비르(darunavir, 2006년 출시)는 암프레나비르 구조를 변형하여 비스-테트라하이드로푸란(THF) 구조를 도입함으로써 약효와 내성 극복 능력을 향상시켰다.[69][60] 이러한 억제제들은 각각의 설계 전략과 특징을 가지며 HIV 치료에 기여해왔다.
5. 1. 결합 부위 및 작용기
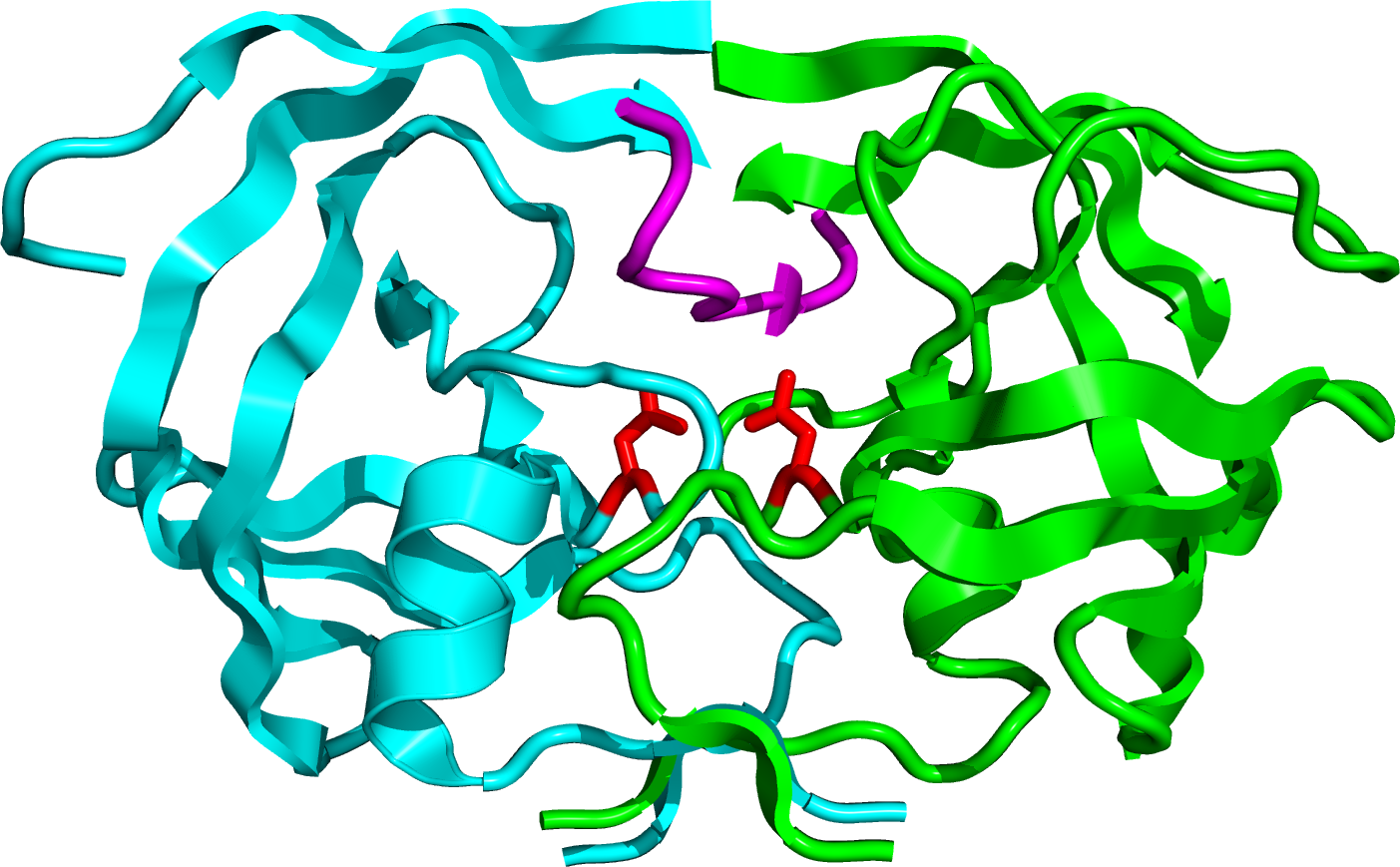
HIV 단백질 분해효소는 99개의 아미노산으로 이루어진 동일한 단량체 2개가 결합한 C2-대칭적 동종이합체 효소이다.[15][6] 각 단량체는 촉매 작용에 필수적인 아스파르트산 잔기인 Asp25와 Asp25'를 활성 부위에 가지고 있다.[6] 이 효소는 Asp-Thr-Gly 서열을 가지는데, 이 서열은 다른 포유류의 아스파르트산 단백질 분해효소에서도 보존되어 나타난다. '플랩(flap)'이라고 불리는 단량체의 확장된 베타 병풍 영역은 기질 결합 부위의 일부를 형성하며, 두 개의 아스파르트산 잔기는 소수성 빈 공간의 바닥에 위치한다.[12][16][17] 각 유연한 플랩은 세 가지 특징적인 영역을 포함한다: 바깥쪽으로 뻗은 곁사슬(Met46, Phe53), 안쪽으로 뻗은 소수성 곁사슬(Ile47, Ile54), 그리고 글리신이 풍부한 영역(Gly48, 49, 51, 52)이다. Ile50은 베타 회전(beta turn)의 끝에 위치하며, 효소에 리간드가 결합하지 않았을 때는 물 분자가 각 단량체의 Ile50 골격과 수소 결합을 형성한다.[17]
HIV 단백질 분해효소는 높은 서열 특이성과 촉매 효율로 펩타이드 결합의 가수분해를 촉매한다. 그 작용 기전은 다른 아스파르트산 단백질 분해효소들과 많은 특징을 공유하지만, 아직 완전히 밝혀지지는 않았다.[12] 물 분자는 플랩의 열리고 닫히는 움직임뿐만 아니라 효소와 기질 사이의 친화력을 높이는 데 중요한 역할을 하는 것으로 보인다. 활성 부위의 아스파르트산 잔기(Asp25, Asp25')는 펩타이드 결합의 가수분해에 직접 관여한다.[17] 이 효소가 선호하는 절단 부위는 프롤린 잔기의 N-말단 쪽이며, 특히 페닐알라닌과 프롤린 사이 또는 타이로신과 프롤린 사이의 결합이다.[6][16]
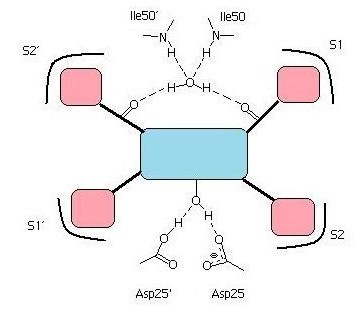
단백질 분해효소 억제제는 효소의 실제 기질이 전이 상태에 있을 때의 구조를 모방하도록 설계되었다. 원래 기질의 펩타이드 결합(–NH-CO–) 대신, 단백질 분해효소에 의해 절단되지 않는 하이드록시에틸렌 그룹(–CH2–CH(OH)–)과 같은 구조를 가진다. 이 억제제들은 HIV 아스파르트산 단백질 분해효소의 활성 부위에 잘 들어맞도록 만들어졌으며, 아스파르트산 단백질 분해효소의 작용 기전에 대한 이해를 바탕으로 설계되었다. 하이드록시에틸아민 구조는 전이 상태를 효과적으로 모방하는 것으로 밝혀졌고, 이는 최초의 단백질 분해효소 억제제인 사퀴나비르의 개발로 이어졌다. 이후 개발된 다른 HIV 단백질 분해효소 억제제들도 대부분 이 원리를 따른다.[15]
HIV 단백질 분해효소 억제제에서 가장 중요한 작용기는 핵심 구조(모티프)에 있는 하이드록시기(–OH)이다. 이 하이드록시기는 기질 결합 부위의 Asp25와 Asp25' 잔기에 있는 카복실산과 수소 결합을 형성한다.[58][70] 펩티도모방체 억제제의 경우, 카보닐기(C=O)가 플랩 부위의 Ile50과 Ile50' 잔기에 연결된 물 분자와 수소 결합을 형성하여 억제제를 플랩 부위에 고정시키는 역할을 한다.[61] 반면, 비펩타이드성 억제제는 양성자 수용체를 가지고 있어, 구조적으로 중요한 물 분자를 대체하고 효소 플랩의 Ile50 잔기와 직접 상호작용한다.[71] HIV 단백질 분해효소의 기질 결합 자리에는 S1, S1', S2, S2'라고 불리는 특정 포켓들이 존재하며, 이들은 기질의 소수성 아미노산을 인식한다. 따라서 억제제가 이 포켓에 맞는 소수성 작용기를 가질 경우 약효가 증가한다.[72] 또한, 억제제의 친수성기는 효소의 기질 결합 부위에 있는 특정 잔기들과 수소 결합을 형성할 수 있다. 예를 들어, 암프레나비르와 다루나비르의 테트라하이드로푸란(THF) 부위가 그러하다. 다루나비르는 암프레나비르의 단일 THF 대신 이중-THF 구조를 가져 더 많은 수소 결합을 형성할 수 있으며, 이는 더 강한 결합 에너지로 이어진다.[69]
5. 2. 약물 개발 과정
단백질 분해효소 억제제는 분해효소의 실제 기질이 전이 상태에 있을 때의 구조를 모방하여 설계되었다. 이는 HIV 아스파르트산 단백질 분해효소의 활성 부위에 잘 맞도록 만들어졌으며, 이 효소의 작용 기전에 대한 지식을 바탕으로 합리적으로 설계되었다.[15] 핵심 원리는 효소가 절단하는 펩타이드 결합(–NH-CO–) 대신, 효소가 절단할 수 없는 하이드록시에틸렌 그룹(-CH2-CH(OH)-) 등으로 대체하는 것이다.[15][57] 여러 구조 중 하이드록시에틸아민(hydroxyethylamine)이 전이 상태를 가장 효과적으로 모방하는 것으로 밝혀졌고, 이는 최초의 단백질 분해효소 억제제인 사퀴나비르(saquinavir)의 발견으로 이어졌다. 이후 개발된 다른 HIV 단백질 분해효소 억제제들도 이와 동일한 원리를 기반으로 설계되었다.[57][15]최초의 HIV 단백질 분해 효소 억제제인 사퀴나비르는 펩티도모방체 하이드록시에틸아민[6]으로, 1995년에 시판되었다.[18] 이는 단백질 분해 효소의 고유 기질의 전이 상태 유사체이다.[6] HIV-1 단백질 분해 효소가 Tyr-Pro 또는 Phe-Pro 서열을 절단한다는 관찰 결과를 바탕으로 설계되었으며,[19] 특히 데카하이드로이소퀴놀린(DIQ) 그룹을 추가하여 수용성과 효능을 향상시킨 것이 중요한 변형이었다.[20] 사퀴나비르는 HIV-1과 HIV-2 모두에 효과적이며[5] 일반적으로 내약성이 좋지만, 혈청에서 높은 농도를 달성하기는 어렵다는 단점이 있다.[11]
리토나비르는 펩티도모방체 HIV 단백질 분해 효소 억제제로 1996년에 시판되었다.[18] 이 약물은 단백질 분해 효소 결합 부위의 C2 대칭성에 맞게 설계되었다.[6] 개발사인 애보트 래버러토리스는 초기 화합물의 낮은 생체 이용률을 개선하기 위해 말단 페닐 잔기를 제거하고 수용성을 높이는 피리딘 그룹을 도입하는 등의 개선을 거쳐 리토나비르를 개발했다.[19] 그러나 상당한 위장관계 부작용과 많은 약 복용량 때문에 단독 치료제로는 잘 사용되지 않는다.[11] 대신, 사이토크롬 P450 효소 대사를 강력하게 억제하는 특성을 이용하여 다른 단백질 분해효소 억제제의 효과를 높이는 약동학적 강화(부스팅) 목적으로 병용 요법에 주로 사용된다.[19][11]
인디나비르 역시 펩티도모방체 하이드록시에틸렌 계열의 HIV 단백질 분해 효소 억제제로, 1996년에 시장에 출시되었다.[6][18] 인디나비르 설계에는 분자 모델링과 억제된 효소 복합체의 X선 결정 구조 분석이 활용되었다. 말단 페닐 구조는 소수성 결합을 통해 효능을 높이는 데 기여한다.[19] 이는 HIV Gag-폴리단백질의 페닐알라닌-프롤린 절단 부위의 유사체이다.[6]
넬피나비르는 펩티도모방체가 아닌 최초의 단백질 분해효소 억제제로, 1997년에 시판되었다.[18] 넬피나비르는 경구 투여가 가능한 비펩티드성 억제제를 목표로 개발되었으며, 기존 펩티드 억제제와 효소의 공동 결정 구조 분석을 통해 억제제의 일부를 비펩티드 치환기로 대체하는 방식으로 설계되었다.[19] 새로운 2-메틸-3-하이드록시벤즈아미드 그룹을 포함하며, C-말단에는 사퀴나비르와 동일한 DIQ 그룹을 가진다.[19] 넬피나비르는 소아 에이즈 치료에 사용이 승인된 최초의 단백질 분해효소 억제제이기도 하다.[19]
앰프레나비르는 1999년에 시장에 출시된[18] ''N'',''N''-이치환 아미노-설폰아미드 계열의 비펩티드 HIV 단백질 분해효소 억제제이다.[6] 사퀴나비르와 유사한 핵심 구조를 가지지만, 양쪽 끝에 테트라하이드로푸란 카바메이트와 이소부틸페닐 설폰아미드라는 다른 작용기를 가지고 있다. 이 구조는 키랄 중심 수를 줄여 합성을 용이하게 하고 수용성을 향상시켜 경구 생체 이용률을 높였다.[19] 그러나 2004년, 여러 면에서 개선된 전구약물인 포삼프레나비르가 출시되면서 시장에서 철수되었다.[6]
로피나비르는 2000년에 시판되었으며,[18] 바이러스의 약물 내성 계통에서 자주 돌연변이가 일어나는 HIV-1 단백질 분해효소의 Val82 잔기와의 상호작용을 줄이도록 설계되었다.[19] 이는 펩티도모방체 HIV 단백질 분해효소 억제제로,[6] 리토나비르와 동일한 핵심 구조를 공유한다. 다만, 리토나비르의 5-티아졸릴 말단 그룹 대신 페녹시아세틸 그룹을 가지며, 2-이소프로필티아졸릴 그룹은 아미노 말단에 6원 고리 요소가 부착된 변형된 발린으로 대체되었다.[19]
포삼프레나비르는 2003년에 시판된[18] 앰프레나비르의 포스포에스테르 전구약물로, 체내에서 빠르고 광범위하게 앰프레나비르로 대사된다.[21] 앰프레나비르보다 용해도와 생체 이용률이 우수하여[6] 하루에 복용해야 하는 알약 수를 줄일 수 있게 되었다.[22]
아타자나비르는 2003년에 시판된[18] 아자펩타이드 계열의 단백질 분해효소 억제제로, 효소 결합 부위의 C2 대칭성에 맞춰 설계되었다.[11] 이전의 HIV 단백질 분해효소 억제제들보다 개선된 내성 프로파일을 보였다.[4] 다른 억제제들과 달리 산성 환경에서만 흡수될 수 있다는 특징이 있다.[11]
티프라나비르는 비펩티드 HIV-1 단백질 분해효소 억제제로,[11] 2005년에 시장에 출시되었다.[18] 다른 억제제들과 달리 비펩티드 쿠마린 구조를 기반으로 개발되었으며, 항단백질 분해효소 활성은 고처리량 스크리닝을 통해 발견되었다.[23] 이 설폰아미드를 포함하는 5,6-디하이드로-4-하이드록시-2-피론 구조는 3-치환 쿠마린 및 디하이드로피론 스크리닝 과정에서 확인되었다.[24] 여러 단백질 분해효소 억제제에 내성을 보이는 HIV-1에 대해서도 광범위한 항바이러스 활성을 가진다.[25]
다루나비르는 2006년에 시장에 출시되었으며,[18] 앰프레나비르의 비펩티드 유사체이다. 가장 큰 특징은 앰프레나비르의 말단 테트라하이드로푸란(THF) 그룹을 두 개의 THF 그룹이 융합된 비스-THF 잔기로 변경한 점이다. 이 구조적 변화는 앰프레나비르보다 더 강력한 효과를 나타내게 하며, 비스-THF 잔기 주변의 입체 화학적 변화는 앰프레나비르에 내성을 가진 단백질 분해효소에도 지속적으로 결합할 수 있도록 한다.[26]
아래는 미국 식품의약국(FDA)의 승인을 받은 HIV 단백질 분해효소 억제제 목록이다.
| 약물 | 구조 |
|---|---|
| 사퀴나비르 |  |
| 넬피나비르 |  |
| 리토나비르 |  |
| 로피나비르 |  |
| 앰프레나비르 |  |
| 포삼프레나비르 |  |
| 다루나비르 |  |
| 인디나비르 |  |
| 아타자나비르 |  |
| 티프라나비르 |  |
5. 3. 구조-활성 관계 (SAR)
시판 중인 대부분의 HIV 단백질분해효소 억제제는 하이드록시에틸렌 골격으로 구성된 중심 코어 모티프를 가지고 있다. 다만, 티프라나비르는 예외적으로 중심 코어가 쿠마린 골격을 기반으로 한다.[15]
HIV 단백질분해효소 억제제에서 구조적으로 매우 중요한 부분은 핵심 모티프에 있는 수산기이다. 이 수산기는 효소의 활성 부위에 위치한 Asp25와 Asp25' 잔기의 카르복실산과 수소 결합을 형성하는데, 이는 억제제의 작용에 핵심적인 역할을 한다.[16][27] 펩티도모방 억제제의 경우, 카르보닐 그룹과 물 분자, 그리고 효소의 플랩(flap) 영역에 있는 Ile50과 Ile50' 잔기 사이에 형성되는 수소 결합이 억제제를 효소에 고정시키는 데 기여하는 것으로 보인다.[19] 반면, 비펩티드 억제제는 4배위 물 분자를 대체하고 효소 플랩의 두 Ile50 잔기와 직접 상호작용하는 양성자 수용체를 가지고 있다.[28]
HIV 단백질분해효소의 결합 부위에는 S1, S1', S2, S2' 등으로 불리는 특정 소수성 포켓이 존재한다. 이 포켓들은 원래 기질의 소수성 아미노산을 인식하도록 되어 있다. 따라서 억제제가 이 소수성 포켓에 잘 맞는 소수성 작용기를 가지고 있을 경우, 효소와의 결합력이 강해져 약효가 증가한다.[29]
또한, 효소 결합 부위의 일부 잔기들은 억제제의 친수성 작용기와 추가적인 수소 결합을 형성할 수 있다. 예를 들어, 암프레나비르와 다루나비르의 테트라하이드로푸란(THF) 부분이 이에 해당한다. 특히 다루나비르는 암프레나비르의 단일 THF 대신 이중-THF(bis-THF) 구조를 가지고 있어 더 많은 수소 결합을 형성할 수 있으며, 이는 더 강한 결합 에너지로 이어진다.[26]
6. HIV 프로테아제 억제제에 대한 내성
HIV는 돌연변이를 통해 단백질 분해효소 억제제에 대한 내성을 획득할 수 있다.[69][26] 이러한 돌연변이는 주로 HIV 프로테아제의 활성 부위에서 발생하지만, 활성 부위 밖에서도 나타난다. 활성 부위 밖의 돌연변이 위치에는 Gag-Pol 폴리펩타이드 전구체의 프로테아제 절단 부위도 포함된다. 절단 부위는 서열이 매우 다양하기 때문에, 프로테아제는 기질을 인식할 때 서열보다는 활성 부위에 결합했을 때 공유하는 보존된 3차원 구조, 즉 '기질 엔벨로프(substrate envelope)'에 의존한다.[73][30]
활성 부위 돌연변이는 주로 억제제가 기질 엔벨로프를 넘어 프로테아제 잔기와 접촉하는 지점에서 발생하여 억제제와의 상호작용을 직접적으로 변화시킨다.[74][31] 반면, 활성 부위가 아닌 곳(비활성 부위)의 돌연변이는 이합체의 안정성이나 단백질의 구조적 유연성에 영향을 미치는 등 다른 메커니즘을 통해 내성을 유발하는 것으로 여겨진다.[75][76][32][33]
지금까지 100개가 넘는 점 돌연변이가 보고되었으며, 그중 적어도 26개는 프로테아제 억제제에 특이적이다. 이 중 약 15개는 약물의 효과를 변화시킬 만큼 중요한 1차 또는 주요 돌연변이로 간주된다.[69][26] 약물 내성을 유발하는 HIV-1 프로테아제의 돌연변이 아미노산 잔기의 예로는 Leu33이 Ile, Val 또는 Phe로, Val82가 Ala, Phe, Leu 또는 Thr로, Ile84가 Val로, Leu90이 Met으로 변하는 것 등이 있다.[77][34]
서로 다른 돌연변이는 각기 다른 프로테아제 억제제에 영향을 미친다. 예를 들어, Leu90에서의 돌연변이는 사퀴나비르(saquinavir)와 넬피나비르(nelfinavir)에 영향을 주지만, 인디나비르(indinavir)의 활성은 Met46, Val82, Ile84에서의 돌연변이에 영향을 받는다. 포삼프레나비르(fosamprenavir)는 Ile50이 Val로 바뀌거나 Ile84에서 변이가 생길 때 영향을 받는다. 여러 부위에 돌연변이가 조합되면 높은 수준의 약물 내성을 나타낼 수 있지만, 단일 돌연변이만으로는 일반적으로 약물 내성이 크지 않다.[69][26]
돌연변이는 1차 돌연변이와 2차 돌연변이로 나눌 수 있다. 1차 돌연변이는 내성에 미치는 영향이 비교적 작다. 하지만 대부분의 프로테아제 억제제는 화학 구조가 유사하기 때문에, 일부 1차 돌연변이가 여러 억제제에 동시에 내성을 유발하는 교차 내성(cross-resistance)을 일으킬 수 있다. 이는 프로테아제 억제제 치료에서 중요한 문제 중 하나이다.[78][35] 지속적인 프로테아제 억제제 치료 과정에서 추가적으로 발생하는 돌연변이를 2차 돌연변이라고 하며, 이는 높은 수준의 약물 내성을 유발할 수 있다.[78][35]
내성 연구를 위해 스탠퍼드 HIV RT 및 프로테아제 서열 데이터베이스(일명 HIV 약물 내성 데이터베이스)가 활용된다. 이 데이터베이스는 1998년에 항레트로바이러스 치료를 받은 환자들의 HIV 역전사 효소 및 프로테아제 서열 정보를 기반으로 구축되었으며, 내성 관련 돌연변이와 유전자형-치료, 유전자형-표현형, 유전자형-결과 간의 상관관계를 분석하는 데 공개적으로 이용 가능하다.[79]
기질 엔벨로프 개념은 기질을 모방하면서도 대부분의 활성 부위 돌연변이에 의한 내성을 피할 수 있도록 엔벨로프 내에 머무르는 억제제를 설계하는 일반적인 전략을 제공한다.[80][81][36][37] 그러나 활성 부위에서 멀리 떨어진 곳의 돌연변이로 인한 약물 내성 문제까지 해결할 수 있는 보편적인 약물 설계 전략은 아직 없다. 따라서 AIDS 치료를 위한 새로운 약물 개발 연구는 기존 약물에 대한 교차 내성을 피하는 데 중점을 두고 진행되고 있다.[53][12]
7. HIV 프로테아제 억제제의 종류 (FDA 승인 기준)
최초의 HIV 단백질 분해 효소 억제제인 사퀴나비르는 펩티도모방체 하이드록시에틸아민[6]으로, 1995년에 시판되었다.[18] 이는 단백질 분해 효소의 고유 기질의 전이 상태 유사체이다.[6] 데카하이드로이소퀴놀린 (DIQ) 그룹의 추가는 사퀴나비르 발견으로 이어진 중요한 변형 중 하나로, 억제제의 입체적 자유도를 제한하여 수용성과 효능을 향상시킨다.[20] 사퀴나비르는 HIV-1과 HIV-2 모두에 효과적이며[5] 일반적으로 내약성이 좋지만, 높은 혈청 농도를 달성하기는 어렵다.[11]
리토나비르는 펩티도모방체 HIV 단백질 분해 효소 억제제로 1996년에 시판되었다.[18] 이는 단백질 분해 효소의 결합 부위에서 C2 대칭에 맞게 설계되었다.[6] 개발 과정에서 생체 이용률이 낮은 초기 화합물을 개선하여 말단 페닐 잔기를 제거하고 물 용해도를 높이기 위해 피리딘 피리딜 그룹을 도입했다.[19] 리토나비르는 상당한 위장관계 부작용과 많은 알약을 복용해야 하는 부담 때문에 단독으로는 잘 사용되지 않는다.[11] 그러나 사이토크롬 P450 효소 매개 대사를 강력하게 억제하므로,[19] 다른 단백질 분해 효소 억제제의 효과를 높이는 약동학적 부스터로 병용 요법에 사용된다.[11]
인디나비르는 펩티도모방체 하이드록시에틸렌 HIV 단백질 분해 효소 억제제로 1996년에 시장에 출시되었다.[6][18] 인디나비르의 설계는 분자 모델링과 억제된 효소 복합체의 X선 결정 구조 분석을 통해 이루어졌다. 말단 페닐 구성 요소는 소수성 결합을 통해 효능을 증가시킨다.[19] 이는 HIV Gag-폴리단백질의 페닐알라닌-프롤린 절단 부위의 유사체이다.[6]
넬피나비르는 펩티도모방체가 아닌 최초의 단백질 분해 효소 억제제로, 1997년에 시판되었다.[18] 넬피나비르는 경구 투여가 가능하고 펩티드 성분이 아닌 억제제를 목표로 설계되었으며, 기존 펩티드 억제제의 단백질 공동 결정 구조 분석을 통해 억제제의 일부를 비펩티드 치환기로 대체하는 방식으로 개발되었다.[19] 새로운 2-메틸-3-하이드록시벤즈아미드 그룹과 사퀴나비르와 동일한 C-말단 DIQ 그룹을 포함한다.[19] 넬피나비르는 소아 에이즈 치료에 사용되도록 지시된 최초의 단백질 분해 효소 억제제였다.[19]
앰프레나비르는 1999년에 시장에 출시된[18] ''N'',''N''-이치환 아미노-설폰아미드 비펩타이드 HIV 단백질 분해 효소 억제제이다.[6] 사퀴나비르와 유사한 핵심 구조를 가지지만, 양쪽 끝에 테트라하이드로푸란 카바메이트 그룹과 아미드가 추가된 이소부틸페닐 설폰아미드라는 다른 작용기를 가지고 있다. 이 구조는 키랄 중심을 줄여 합성을 용이하게 하고 수용성을 향상시켜 더 나은 경구 생체 이용률을 제공한다.[19] 그러나 2004년, 여러 면에서 더 우수한 전구약물인 포삼프레나비르가 출시되면서 시장에서 철수되었다.[6]
로피나비르는 2000년에 시판되었으며,[18] 바이러스의 약물 내성 계통에서 자주 돌연변이되는 HIV-1 단백질 분해 효소의 Val82 잔기와의 상호 작용을 줄이도록 설계되었다.[19] 이는 펩티도모방체 HIV 단백질 분해 효소 억제제로,[6] 리토나비르와 동일한 핵심 구조를 가진다. 리토나비르의 5-티아졸릴 말단 그룹 대신 페녹시아세틸 그룹을 가지며, 2-이소프로필티아졸릴 그룹은 아미노 말단에 6원 고리 요소가 부착된 변형된 발린으로 대체되었다.[19]
포삼프레나비르는 2003년에 시판되었으며,[18] 체내에서 빠르고 광범위하게 앰프레나비르로 대사되는 포스포에스테르 전구약물이다.[21] 앰프레나비르보다 용해도와 생체 이용률이 우수하여[6] 하루에 복용해야 하는 알약의 수를 줄일 수 있다.[22]
아타자나비르는 2003년에 시판된[18] 아자펩타이드 단백질 분해 효소 억제제로, 효소 결합 부위의 C2 대칭에 맞게 설계되었다.[11] 이전의 HIV 단백질 분해 효소 억제제보다 개선된 내성 프로파일을 보였다.[4] 다른 단백질 분해 효소 억제제와 달리 산성 환경에서만 흡수된다는 특징이 있다.[11]
티프라나비르는 비펩티드 HIV-1 단백질 분해 효소 억제제로,[11] 2005년에 시장에 출시되었다.[18] 다른 HIV 단백질 분해 효소 억제제와 달리 비펩티드 쿠마린 템플릿에서 개발되었으며, 항단백질 분해 효소 활성은 고처리량 스크리닝을 통해 발견되었다.[23] 이 설폰아미드 함유 5,6-디하이드로-4-하이드록시-2-피론 구조는 3-치환 쿠마린 및 디하이드로피론 스크리닝 과정에서 확인되었다.[24] 여러 단백질 분해 효소 억제제에 내성을 보이는 HIV-1에 대해 광범위한 항바이러스 활성을 가진다.[25]
다루나비르는 2006년에 시장에 출시되었으며,[18] 앰프레나비르의 비펩티드 유사체이다. 중요한 구조적 변화는 말단 테트라하이드로푸란(THF) 그룹에 있는데, 단일 THF 그룹 대신 두 개의 THF 그룹이 융합된 비스-THF 잔기를 포함하여 앰프레나비르보다 더 효과적이다. 이 비스-THF 잔기 주변의 입체 화학적 배열은 앰프레나비르에 내성을 갖게 된 단백질 분해 효소와도 지속적으로 결합할 수 있도록 한다.[26]
모든 FDA 승인 단백질 분해 효소 억제제는 다음과 같다.
8. 최근 동향 및 미래 전망
2018년 1월 기준으로 다루나비르(darunavir)는 시판된 가장 최신 HIV 단백질분해효소 억제제이다.[38] 하지만 새로운 약물 개발이 계속 시도되고 있다. 예를 들어, 2006년 글락소스미스클라인(GlaxoSmithKline)은 제형 관련 문제로 인해 개발 중이던 브레카나비르(brecanavir)의 2상 임상 개발을 중단한 바 있다.[39]
한편, 약물 내성 문제를 극복하기 위한 새로운 접근법도 모색되고 있다. 2009년 여름, 글락소스미스클라인과 콘서트 제약(Concert Pharmaceuticals)은 중수소를 포함하는 의약품 개발 협력을 발표했다.[40] 이 중 하나인 CTP-518은 아타자나비르(atazanavir)의 특정 수소 원자를 중수소로 대체하여 개발된 새로운 HIV 단백질분해효소 억제제이다. CTP-518은 2009년 하반기에 1상 임상 시험에 들어갈 것으로 예상되었다.[40] 전임상 연구 결과, 이러한 중수소 치환은 항바이러스 효능을 유지하면서도 간 대사를 늦춰 약물의 반감기와 혈장 최저 농도를 증가시킬 수 있는 것으로 나타났다. 이는 CTP-518이 리토나비르(ritonavir)와 같은 약효 증강제 없이 단독으로 사용될 수 있는 최초의 HIV 단백질분해효소 억제제가 될 가능성을 시사한다.[40]
HIV의 약물 내성 돌연변이는 주로 단백질분해효소의 활성 부위나 그 주변에서 발생하며, 약물과의 상호작용을 변화시킨다.[26][31] 연구자들은 바이러스가 기질을 인식하는 3차원적 형태인 '기질 엔벨로프(substrate envelope)' 개념을 활용하여, 이 엔벨로프 내에 머무르도록 억제제를 설계함으로써 일부 활성 부위 돌연변이로 인한 내성을 피하려는 전략을 사용하고 있다.[36][37] 그러나 모든 종류의 내성 문제, 특히 활성 부위에서 멀리 떨어진 돌연변이에 의한 내성을 해결할 수 있는 일반적인 전략은 아직 없다. 따라서 현재 AIDS 치료 연구는 기존 약물에 대한 교차 내성을 피하는 새로운 치료법 개발에 집중하고 있다.[12]
참조
[1]
논문
Natural occurring polyphenols as template for drug design. Focus on serine proteases
[2]
논문
Synthesis and SAR studies of potent HIV protease inhibitors containing novel dimethylphenoxyl acetates as P2 ligands
[3]
논문
Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography
[4]
논문
Molecular basis for increased susceptibility of isolates with atazanavir resistance-conferring substitution I50L to other protease inhibitors
[5]
논문
Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use
[6]
서적
Goodman and Gilmans's The Pharmacological Basis of Therapeutics
McGraw-Hill
[7]
EMedicine
HIV Disease
[8]
논문
HIV-1 Protease Inhibitors: A Comparative QSAR Analysis
[9]
논문
Expedient solid-phase synthesis of both symmetric and asymmetric diol libraries targeting aspartic proteases
[10]
논문
Targeting proteases: successes, failures and future prospects
[11]
웹사이트
HIV protease inhibitors
http://www.uptodate.[...]
UpToDate
2014-06-17
[12]
문서
HIV-1 protease: mechanism and drug discovery
2003
[13]
문서
Antiretroviral drugs
2007
[14]
문서
HIV protease inhibitor-specific alterations in human adipocyte differentiation and metabolism
2006
[15]
문서
The history of antiretrovirals: key discoveries over the past 25 years
2009
[16]
문서
Structure-activity relationship of orally potent tripeptide-based HIV protease inhibitors containing hydroxymethylcarbonyl isostere
2000
[17]
문서
Drug design: New inhibitors for HIV-1 protease based on Nelfinavir as lead
2007
[18]
문서
HIV drug development: the next 25 years
2007
[19]
문서
Rational approach to AIDS drug design through structural biology
2002
[20]
서적
Enzymes and Their Inhibition: Drug Development
CRC press
2005
[21]
문서
Steady-state pharmacokinetics of once-daily fosamprenavir/ritonavir and atazanavir/ritonavir alone and in combination with 20mg omeprazole in healthy volunteers
2007
[22]
문서
Fosamprenavir – A Review of its Use in the Management of Antiretroviral Therapy-naive Patients with HIV Infection
2004
[23]
문서
Tipranavir inhibits broadly protease inhibitor-resistant HIV-1 clinical samples
2000
[24]
문서
Tipranavir analogous 3-sulfonylanilidotetronic acids: new synthesis and structure-dependent anti-HIV activity
2008
[25]
문서
Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir
2005
[26]
문서
Darunavir: A nonpeptidic antiretroviral protease inhibitor
2007
[27]
문서
Effect of Flap Mutations on Structure of HIV-protease and Inhibition by Saquinavir and Darunavir
2008
[28]
문서
Approaches to the Design of Effective HIV-1 Protease Inhibitors
2000
[29]
문서
Achiral oligoamines as versatile tool for the development of aspartic protease inhibitors
2008
[30]
문서
Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes
2002
[31]
문서
Combating susceptibility to drug resistance: lessons from HIV-1 protease
2004
[32]
문서
Resistance mechanism revealed by crystal structures of unliganded nelfinavir-resistant HIV-1 protease non-active site mutants N88D and N88S
2009
[33]
문서
Elucidating a relationship between conformational sampling and drug resistance in HIV-1 protease
2013
[34]
서적
Foye´s Principles of Medicinal Chemistry
Lippincott williams & Wilkins, a Wolters Kluwer business
2008
[35]
서적
Antiretroviral Resistance in Clinical Practice
Mediscript Ltd
2008
[36]
논문
Toward the design of mutation-resistant enzyme inhibitors: further evaluation of the substrate envelope hypothesis
2009-09
[37]
논문
Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance
2010-05
[38]
논문
Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV
[39]
웹사이트
GlaxoSmithKline discontinues clinical development of investigational protease inhibitor brecanavir (640385)
http://www.gsk.com/m[...]
2008-06-11
[40]
웹사이트
GSK and Concert Pharmaceuticals form alliance to develop novel deuterium-modified drugs
http://www.gsk.com/m[...]
2009-11-05
[41]
저널
Natural occurring polyphenols as template for drug design. Focus on serine proteases
[42]
저널
Synthesis and SAR studies of potent HIV protease inhibitors containing novel dimethylphenoxyl acetates as P2 ligands
[43]
저널
Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography
[44]
저널
Molecular basis for increased susceptibility of isolates with atazanavir resistance-conferring substitution I50L to other protease inhibitors
https://archive.org/[...]
[45]
저널
Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use
[46]
서적
Goodman and Gilmans's The Pharmacological Basis of Therapeutics
https://archive.org/[...]
McGraw-Hill
[47]
EMedicine
HIV Disease
[48]
서적
Goodman and Gilmans's The Pharmacological Basis of Therapeutics
https://archive.org/[...]
McGraw-Hill
[49]
저널
HIV-1 Protease Inhibitors: A Comparative QSAR Analysis
[50]
저널
Expedient solid-phase synthesis of both symmetric and asymmetric diol libraries targeting aspartic proteases
[51]
저널
Targeting proteases: successes, failures and future prospects
[52]
웹인용
HIV protease inhibitors
http://www.uptodate.[...]
UpToDate
2014-06-17
[53]
논문
HIV-1 protease: mechanism and drug discovery
[54]
논문
Antiretroviral drugs
[55]
논문
HIV-1 protease: mechanism and drug discovery
[56]
논문
HIV protease inhibitor-specific alterations in human adipocyte differentiation and metabolism
[57]
논문
The history of antiretrovirals: key discoveries over the past 25 years
[58]
논문
Structure-activity relationship of orally potent tripeptide-based HIV protease inhibitors containing hydroxymethylcarbonyl isostere
[59]
논문
Drug design: New inhibitors for HIV-1 protease based on Nelfinavir as lead
[60]
논문
HIV drug development: the next 25 years
[61]
논문
Rational approach to AIDS drug design through structural biology
[62]
서적
Enzymes and Their Inhibition: Drug Development (6th edition)
CRC press
[63]
저널
Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use
[64]
논문
Steady-state pharmacokinetics of once-daily fosamprenavir/ritonavir and atazanavir/ritonavir alone and in combination with 20mg omeprazole in healthy volunteers
[65]
논문
Fosamprenavir – A Review of its Use in the Management of Antiretroviral Therapy-naive Patients with HIV Infection
[66]
논문
Tipranavir inhibits broadly protease inhibitor-resistant HIV-1 clinical samples
[67]
논문
Tipranavir analogous 3-sulfonylanilidotetronic acids: new synthesis and structure-dependent anti-HIV activity
[68]
논문
Selection and characterization of HIV-1 showing reduced susceptibility to the non-peptidic protease inhibitor tipranavir
[69]
논문
Darunavir: A nonpeptidic antiretroviral protease inhibitor
[70]
논문
Effect of Flap Mutations on Structure of HIV-protease and Inhibition by Saquinavir and Darunavir
[71]
논문
Approaches to the Design of Effective HIV-1 Protease Inhibitors
2000
[72]
논문
Achiral oligoamines as versatile tool for the development of aspartic protease inhibitors
2008
[73]
논문
Substrate shape determines specificity of recognition for HIV-1 protease: analysis of crystal structures of six substrate complexes
2002
[74]
논문
Combating susceptibility to drug resistance: lessons from HIV-1 protease
2004-10
[75]
논문
Resistance mechanism revealed by crystal structures of unliganded nelfinavir-resistant HIV-1 protease non-active site mutants N88D and N88S
2009
[76]
논문
Elucidating a relationship between conformational sampling and drug resistance in HIV-1 protease
2013-05-14
[77]
서적
Foye´s Principles of Medicinal Chemistry
Lippincott williams & Wilkins, a Wolters Kluwer business
2008
[78]
서적
Antiretroviral Resistance in Clinical Practice
Mediscript Ltd
2008
[79]
저널
Rationale and Uses of a Public HIV Drug-Resistance Database
[80]
논문
Toward the design of mutation-resistant enzyme inhibitors: further evaluation of the substrate envelope hypothesis
2009-09
[81]
논문
Evaluating the substrate-envelope hypothesis: structural analysis of novel HIV-1 protease inhibitors designed to be robust against drug resistance
2010-05
[82]
논문
Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV
2009
[83]
웹인용
Archived copy
http://www.gsk.com/m[...]
2008-06-11
[84]
웹인용
Archived copy
http://www.gsk.com/m[...]
2009-11-05
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.
문의하기 : help@durumis.com