비교유전체학
"오늘의AI위키"의 AI를 통해 더욱 풍부하고 폭넓은 지식 경험을 누리세요.
1. 개요
비교유전체학은 여러 생물의 게놈을 비교하여 진화적 관계, 유전자 기능, 유전체 구조 등을 연구하는 학문이다. 1980년대 초 바이러스 게놈 비교에서 시작되어, 1990년대 중반 이후 게놈 서열 분석 기술 발전과 함께 급속도로 발전했다. 진화론을 이론적 기반으로 하여, 게놈 서열의 유사성을 분석하고, 상동성, 선택, 복제수 변이 등을 연구한다. 서열 정렬, 전체 게놈 정렬, 계통 발생 재구성, 유전자 지도 작성 등의 방법론을 사용하며, UCSC 브라우저, Ensembl, VISTA 등 다양한 도구들이 활용된다. 의학 연구, 농업, 생물학 연구 등 다양한 분야에 응용되어 질병 관련 유전자 발굴, 백신 개발, 작물 개량 등에 기여한다.

더 읽어볼만한 페이지
2. 역사
비교유전체학은 1980년대 초 바이러스 게놈 비교에서 시작되었다.[20] 예를 들어, 동물을 감염시키는 작은 RNA 바이러스(피코르나바이러스)와 식물을 감염시키는 바이러스(카우피 모자이크 바이러스)를 비교한 결과, 상당한 서열 유사성과 일부 유전자 순서의 유사성을 보였다.[23] 1986년, 100개 이상의 유전자를 가진 수두 대상 포진 바이러스와 엡스타인-바 바이러스의 게놈을 비교하는 최초의 대규모 비교 유전체학 연구가 발표되었다.[24]
진화론은 비교유전체학의 이론적 기반이며, 동시에 비교유전체학의 결과는 진화론을 전례 없이 풍부하게 하고 발전시켰다.[32][33] 둘 이상의 게놈 서열을 비교하면 계통수에서 서열의 진화적 관계를 추론할 수 있다. 다양한 생물학적 게놈 데이터와 수직 및 수평 진화 과정을 연구하여 유전자 구조와 조절 기능의 중요한 부분을 이해할 수 있다.[32][33]
1995년 ''인플루엔자균'' Rd의 게놈 서열이 발표되면서 세포 생물의 첫 번째 완전한 게놈 서열이 밝혀졌다.[25] 같은 해에 작은 기생 박테리아 ''미코플라스마 제니탈리움''의 게놈 서열 분석 논문이 발표되었다.[26]
1998년 Art Delcher, Simon Kasif 및 Steven Salzberg는 MUMMER 시스템을 개발하여 미생물 게놈 비교 연구를 가속화했다. MUMMER는 대규모 재배열, 단일 염기 돌연변이, 반전, 탠덤 반복 확장 및 기타 다형성을 식별하는 데 사용되었다.[20]
최초의 진핵생물 게놈 서열은 1996년에 발표된 빵 효모 ''사카로마이세스 세레비지애''였다.[27] 이후 1998년에는 선충 ''예쁜꼬마선충'',[15] 2000년에는 초파리 ''초파리'' 게놈이 해독되었다.[29] 제럴드 M. 루빈과 그의 팀은 ''초파리'', ''예쁜꼬마선충'', ''사카로마이세스 세레비지애'' 진핵생물의 게놈과 원핵생물 ''인플루엔자균''의 게놈을 비교하는 논문을 발표했다.[29]
2000년대에는 인간, 참복 ''참복'', 생쥐 등 척추동물의 대규모 게놈이 발표되면서, 대규모 게놈 비교 연구가 가능해졌다.[31]
2007년 차세대 염기 서열 분석 방법이 도입되면서 방대한 양의 게놈 데이터가 생성되었고, 비교유전체학 연구가 폭발적으로 증가했다.[21]
3. 진화적 원리
관련된 게놈의 유사성은 비교유전체학의 기초이다. 두 생물이 최근 공통 조상을 가지고 있다면 두 종의 게놈 간의 차이점은 조상의 게놈에서 진화한 것이다. 두 유기체의 관계가 가까울수록 게놈 간의 유사성이 높아진다. 두 유기체 간에 밀접한 관계가 있다면 게놈은 공통염색체 거동을 나타내는데, 즉 유전자 서열의 일부 또는 전체가 보존된다. 따라서 게놈 서열은 알려진 기능의 유전자와 상동성(서열 유사성)을 분석하여 유전자 기능을 식별하는 데 사용할 수 있다.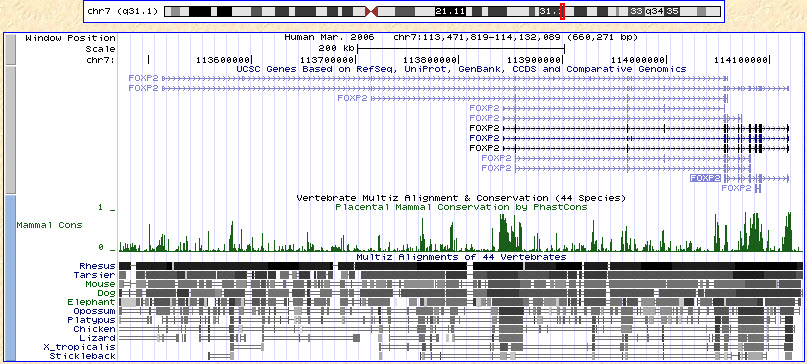
상동 서열은 다른 종의 관련 서열이다. 원래 종에 유전자가 존재하고, 그 종이 두 종으로 나뉘어지면 새로운 종의 유전자는 원래 종의 서열과 상동성을 갖는다. 상동 유전자 서열은 유전자 복제(유전자 중복)에 의해 분리된다. 게놈의 특정 유전자가 복사되면 두 서열의 복사본은 원래 유전자와 상동성을 갖는다. 상동 서열 쌍은 상동 유전자 쌍(orthologs)이라고 하고, 상동 유전자 서열 쌍은 측계 유전자 쌍(paralogs)이라고 한다. 상동 유전자 쌍은 일반적으로 동일하거나 유사한 기능을 가지며, 측계 유전자 쌍의 경우는 그렇지 않을 수 있다. 측계 유전자 쌍에서 서열은 다른 기능을 갖도록 진화하는 경향이 있다.
비교유전체학은 다양한 유기체의 단백질, RNA, 조절 영역의 유사점과 차이점을 모두 활용하여 선택이 이러한 요소에 어떻게 작용했는지 추론한다. 다른 종 간의 유사성을 담당하는 요소는 시간이 지남에 따라 보존되어야 하며(안정화 선택), 종 간의 차이를 담당하는 요소는 발산해야 한다(양성 선택). 마지막으로, 유기체의 진화적 성공에 중요하지 않은 요소는 보존되지 않는다(선택은 중립적이다).
이 분야의 중요한 목표 중 하나는 진핵생물 게놈 진화의 메커니즘을 식별하는 것이다. 그러나 개별 계통의 역사 전반에 걸쳐 발생한 다양한 사건으로 인해 종종 복잡해지며, 각 생물의 게놈에 왜곡되고 중첩된 흔적만 남게 된다. 이러한 이유로 작은 모델 유기체 연구는 진화의 일반적인 메커니즘에 대한 이해를 발전시키는 데 매우 중요하다.[32][33]
3. 1. 상동성
3. 2. 선택
3. 3. 복제수 변이 (CNV)
복제수 변이(Copy Number Variation, CNV)는 DNA의 큰 부분의 유전자 삭제 또는 유전자 중복을 포함하며, 유전적 다양성의 주요 원인으로 인식된다.[34] CNV는 유전자 구조, 유전자 용량, 유전자 발현 조절에 영향을 미친다. 단일 염기 다형성(SNP)이 더 흔하지만, CNV는 더 큰 유전체 영역에 영향을 미치며 표현형과 다양성에 심오한 영향을 미칠 수 있다.[34] 인간 게놈의 약 4.8~9.5%를 차지하며 상당한 기능적, 진화적 영향을 미친다.[35]
포유류에서 CNV는 유전자 발현과 다양한 표현형질에 영향을 미치며, 개체군 다양성에 크게 기여한다.[35] 인간과 침팬지 게놈을 비교 분석한 결과, CNV가 단일 뉴클레오티드 변화에 비해 진화적 변화에 더 큰 역할을 할 수 있음이 밝혀졌다. CNV는 개별 염기쌍 변화보다 더 많은 뉴클레오티드에 영향을 미치며, SNP가 1.2%인 것에 비해 CNV는 게놈의 약 2.7%에 영향을 미친다. 많은 CNV가 인간과 침팬지 사이에 공유되지만, 상당 부분은 각 종에 고유하다.[36]
CNV는 인간의 유전 질환과 관련이 있어, 인간 건강에서의 중요성을 강조한다. CNV의 기원과 진화적 적응 및 질병에 대한 기여를 포함하여, CNV에 대한 많은 질문이 아직 해결되지 않았다. 지속적인 연구는 비교 게놈 혼성화와 같은 기술을 사용하여 이러한 질문에 답하려 하며, 이를 통해 CNV와 그 중요성에 대한 자세한 조사가 가능하다.[36]
4. 방법론
유전체에 담긴 방대한 양의 데이터를 고려할 때, 유전체 비교에는 계산적 접근 방식이 필수적이다. 현재 전유전체 비교부터 유전자 발현 분석에 이르기까지 다양한 도구들이 공개적으로 사용 가능하다.[43] 여기에는 시스템 및 제어, 정보 이론, 문자열 분석 및 데이터 마이닝의 접근 방식이 포함된다.[44] 특히 정보 과학과 유전체 생물학을 함께 가르칠 때, 계산적 접근 방식은 연구와 교육에 매우 중요하게 사용될 것이다.[45]
비교 유전체학은 유전체 크기와 유전자 밀도의 기본적인 비교로 시작한다. 예를 들어, 유전체 크기는 코딩 능력과 잠재적으로 조절 이유로 중요하다. 높은 유전자 밀도는 유전체 주석, 환경 선택 분석을 용이하게 한다. 반대로, 낮은 유전자 밀도는 인간 유전체에서와 같이 유전 질환의 매핑을 방해한다.
4. 1. 서열 정렬
서열 정렬은 유전체 서열 간 유사성을 비교하는 기본적인 방법으로, 계통, 공통 진화적 기원, 또는 공통 구조 및 기능과 같은 유사한 서열에 대한 정보를 얻는데 사용된다.[47][48] 정렬은 뉴클레오티드와 단백질 서열 모두에 대해 수행할 수 있다.[47][48] 정렬은 지역 또는 전역 쌍별 정렬, 그리고 다중 서열 정렬로 구성된다. 전역 정렬은 니들만-분쉬 알고리즘으로 알려진 동적 프로그래밍 알고리즘을 사용하여 찾을수 있다. 스미스-워터만 알고리즘은 지역 정렬을 찾는 데 사용된다. 서열 데이터베이스의 기하급수적인 성장과 더 긴 서열의 출현으로 인해 더 빠르고, 근사적이며, [https://science.umd.edu/labs/delwiche/bsci348s/lec/AlignHeuristic.html#:~:text=Heuristic%20Alignment&text=BLAST%20is%20a%20pairwise%20local,sequence%20databases%20such%20as%20GenBank. 발견적 정렬] 절차에 대한 관심이 높아지고 있다. 이들 중 '''FASTA'''와 '''BLAST''' 알고리즘은 지역 쌍별 정렬에 두드러진다.최근 몇 년 동안 '''MUMmer'''(1999), '''BLASTZ'''(2003), '''AVID'''(2003)와 같이 긴 서열을 정렬하도록 맞춤화된 프로그램이 개발되었다. BLASTZ는 지역적 접근 방식을 채택하는 반면, MUMmer와 AVID는 전역 정렬을 지향한다. 지역 및 전역 정렬 접근 방식의 이점을 모두 활용하기 위해, 한 가지 효과적인 전략은 이를 통합하는 것이다. 처음에는 '''BLAT'''으로 알려진 BLAST의 빠른 변형을 사용하여 상동 "앵커" 영역을 식별한다. 이러한 앵커는 보존된 순서와 방향을 나타내는 세트를 식별하기 위해 후속적으로 정밀 검사를 받는다. 그런 다음 이러한 앵커 세트는 전역 전략을 사용하여 정렬된다.[49][50]또한, 진행 중인 노력은 속도를 향상시켜 방대한 양의 유전체 서열 데이터를 처리하도록 기존 알고리즘을 최적화하는 데 중점을 둔다. 또한 '''MAVID'''는 여러 유전체를 정렬하도록 특별히 설계된 또 다른 주목할 만한 쌍별 정렬 프로그램이다.
유전체 서열 데이터의 쌍별 비교는 비교 유전자 예측에서 널리 활용된다. 비교 기능 유전체학의 많은 연구는 쌍별 비교에 의존하며, 여기서 각 유전자의 특성은 종 전반의 다른 유전자의 특성과 비교된다. 이 방법은 고유한 관찰보다 훨씬 더 많은 비교를 생성하여 각 비교를 다른 비교에 종속시킨다.[49][50]
여러 유전체의 비교는 쌍별 종간 비교의 자연스러운 확장이다. 이러한 비교는 일반적으로 두 개의 계통 발생적 척도에서 보존된 영역을 식별하는 것을 목표로 한다. 1. '''계통 발생적 발자국 찍기'''[51]라고도 하는 심층 비교는 척추 동물과 같은 더 높은 분류 단위에서 보존을 밝혀낸다.[52] 2. 최근에 '''계통 발생적 그림자 추적''',[53]이라고 불리는 얕은 비교는 밀접하게 관련된 종 그룹에서 보존을 조사한다.
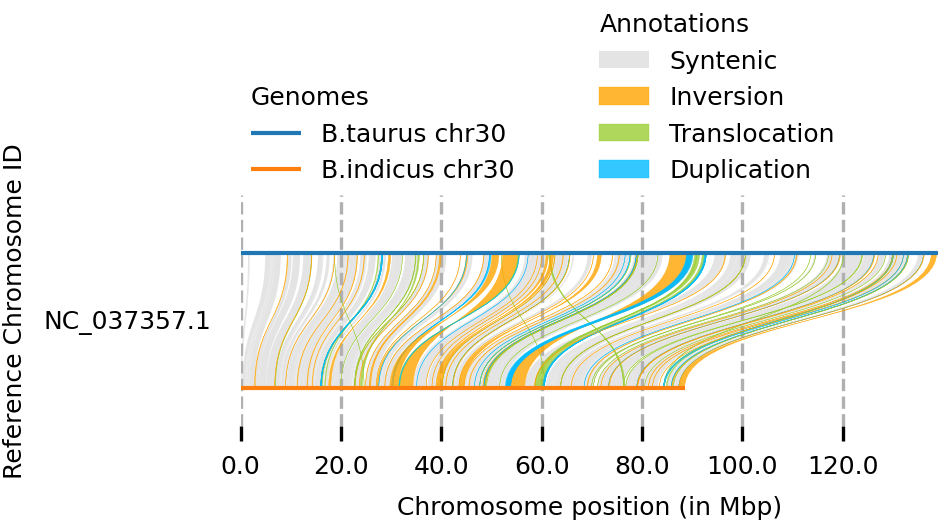
4. 2. 전체 게놈 정렬 (WGA)
전체 게놈 정렬(Whole-Genome Alignment, WGA)은 둘 이상의 게놈 사이의 뉴클레오티드 수준에서 진화 관계를 예측하는 방법이다.[54] 이는 선형 서열 정렬과 유전자 직교 예측의 요소를 통합하며, 전체 게놈의 방대한 크기와 복잡한 특성으로 인해 더 큰 과제를 제시한다. WGA는 계통 발생 추론, 게놈 주석 및 기능 예측과 같은 다양한 게놈 전체 분석에서 중요한 역할을 한다.[54] SyRI(Synteny and Rearrangement Identifier, 구문론 및 재배열 식별자)는 전체 게놈 조립체 간의 구조적 및 서열 차이를 모두 식별하도록 설계된 도구이다.[55] SyRI는 WGA를 입력으로 받아 게놈 구조의 불일치를 검색하고, 재배열된 영역과 재배열되지 않은(동일한) 영역 내에서 국소 서열 변이를 식별한다.[55]
4. 3. 계통 발생 재구성
계통 발생 재구성은 공통 조상의 관점에서 진화적 관계를 설명하는 데 사용된다. 이러한 관계는 일반적으로 계통수라고 하는 트리로 표현된다.[56] 응집 이론은 개체군 내의 유전자의 대립 유전자를 개체군 구성원이 공유하는 단일 조상 복사본으로 추적하는 소급적 모델이다. 이것은 또한 최근 공통 조상으로 알려져 있다. 응집 이론에 기초한 분석은 돌연변이의 도입과 개체군 내 특정 대립 유전자 또는 유전자 분포 사이의 시간을 예측하려고 시도한다. 이 기간은 최근 공통 조상이 존재했던 시간과 같다. 유전 관계는 계통수와 유사한 형태로 시각화된다. 응집 (또는 유전자 계보)은 덴드로그램을 사용하여 시각화할 수 있다.[56]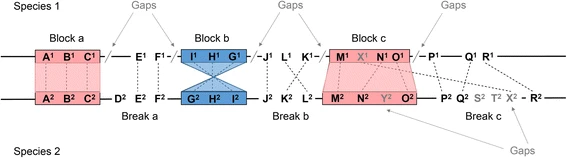
4. 4. 유전자 지도 작성
유전자 지도 작성에서 상동성 시각화는 염색체에서 보존된 유전자 순서를 확인하는 한 가지 방법이다.[58] 이는 일반적으로 공통 조상으로부터 유래된 관련 종의 염색체에 사용된다.[58] 이 방법과 다른 방법들은 진화 역사를 밝히는 데 도움이 될 수 있다. 최근 연구에서는 비교유전체학을 사용하여 포유류 계통발생학 전반에 걸쳐 16개의 조상 핵형을 재구성했다. 전산 재구성은 포유류 진화 과정에서 염색체가 어떻게 재배열되었는지 보여주었다. 이는 종종 발달 과정의 조절과 관련된 선택적 영역의 보존에 대한 통찰력을 제공했다. 또한 염색체 진화와 DNA 재배열과 관련된 유전 질환에 대한 이해를 제공하는 데 도움이 되었다.
5. 도구
게놈 데이터의 방대한 가용성으로 인해 서열 및 전체 게놈 분석을 위한 전산 도구가 빠르게 개발되고 있으며, 비교 분석 도구 또한 발전하고 개선되고 있다.[59] 이러한 분석 과제에서 비교 결과를 시각화하는 것은 매우 중요하다.[59] 긴 게놈 영역의 정렬을 수동으로 검사하는 것은 매우 비효율적이기 때문에, 인터넷 기반 게놈 브라우저는 게놈 영역에 대한 모든 서열 기반 생물학적 정보를 통합하여 게놈 서열을 조사하기 위한 많은 유용한 도구를 제공한다.[59] 이는 대량의 관련 생물학적 데이터를 추출할 때 사용하기 매우 쉽고 시간을 절약할 수 있게 해준다.[59]
- '''UCSC 브라우저''': 대규모 게놈 컬렉션에 대한 참조 서열 및 작업 초안 어셈블리를 포함한다.[60]
- '''Ensembl''': 척추 동물 및 기타 진핵생물 종에 대한 게놈 데이터베이스를 생성하고 이 정보를 온라인에서 자유롭게 사용할 수 있도록 한다.[61]
- '''MapView''': 광범위한 게놈 매핑 및 서열 데이터를 제공한다.[62]
- '''VISTA''': 게놈 서열의 비교 분석을 위한 포괄적인 프로그램 및 데이터베이스 모음이다. DNA 정렬을 기반으로 비교 분석 결과를 시각화하기 위해 구축되었으며, 소규모 및 대규모 데이터 모두에 쉽게 적합할 수 있다.[63]
- '''BlueJay 게놈 브라우저''': 주석이 달린 게놈 및 기타 게놈 요소의 다중 스케일 보기를 위한 독립형 시각화 도구이다.[64]
- '''[https://schneebergerlab.github.io/syri/ SyRI]''': 전장 게놈 어셈블리(WGA)를 사용하여 관련 게놈 간의 게놈 차이를 예측하는 데 도움이 되는 합성 분석 및 시각화 기능을 제공한다.[65]
- '''[https://genomevolution.org/wiki/index.php/SynMap2 Synmap2]''': 유전자 지도 또는 어셈블리를 효율적으로 비교하여 관련 유기체 간의 게놈 진화 및 재배열에 대한 통찰력을 제공한다.[66]
- '''[https://github.com/hsinnan75/GSAlign GSAlign]''': 게놈 서열의 정확한 정렬을 용이하게 하며, 특히 대규모 비교 유전체학 연구에 유용하여 연구자가 게놈 전체의 유사점과 차이점을 식별할 수 있다.[67]
- '''[https://igv.org/ IGV (통합 유전체학 뷰어)]''': 게놈 데이터를 시각화하고 분석하기 위해 널리 사용되는 도구로, 사용자가 여러 게놈에서 정렬, 변형 및 주석을 탐색할 수 있도록 하여 비교 유전체학을 지원한다.[68]
- '''[https://github.com/Illumina/manta Manta]''': 삽입, 삭제, 반전 및 중복과 같은 게놈 재배열을 감지하여 개체군 또는 종 간의 유전적 변이를 이해하는 데 도움이 되는 비교 유전체학에 중요한 신속한 구조적 변이 호출자이다.[69]
- '''[https://bioinformaticstools.mayo.edu/research/cnvnator/ CNVNatar]''': 게놈 진화 및 개체군 유전학을 이해하는 데 중요한 복사본 수 변이(CNV)를 감지하는 데 특화되어 있어 다양한 유기체 간의 게놈 구조적 변화에 대한 통찰력을 제공한다.[70]
- '''[http://pipmaker.bx.psu.edu/pipmaker/ PIPMaker]''': 두 게놈 서열의 정렬 및 비교를 용이하게 하여 보존된 영역, 중복 및 진화적 중단점을 식별하여 비교 유전체학 분석을 지원한다.[71]
- '''[https://cb.csail.mit.edu/cb/glass/ GLASS (전체 게놈 위치 및 서열 검색기)]''': 유전자 조절 및 진화에 대한 이해에 초점을 맞춘 비교 유전체학 연구에 중요한 게놈 전체에서 보존된 조절 요소를 식별하는 도구이다.[72]
- '''PatternHunter''': 게놈 서열 전체에서 보존된 패턴, 모티프 및 반복을 식별하는 기능을 제공하는 다목적 서열 분석 도구로, 유전자족 및 조절 요소의 비교 유전체학 연구를 지원한다.
- '''[https://github.com/mummer4/mummer Mummer]''': 전체 게놈 정렬 및 비교를 위한 도구 모음으로, 다양한 규모의 게놈 간의 유사점, 차이점 및 진화적 사건을 식별하기 위해 비교 유전체학에서 널리 사용된다.[73]
온라인 도구를 사용하면 이러한 웹사이트가 지속적으로 개발 및 업데이트된다는 장점이 있다. 효율성을 향상시키기 위해 온라인에서 사용할 수 있는 많은 새로운 설정과 콘텐츠가 있다.[59]
5. 1. 게놈 브라우저
게놈 데이터의 방대한 가용성으로 인해 서열 및 전체 게놈 분석을 위한 전산 도구가 빠르게 개발되고 있으며, 비교 분석 도구 또한 발전하고 개선되고 있다.[59] 이러한 분석 과제에서 비교 결과를 시각화하는 것은 매우 중요하다.[59] 긴 게놈 영역의 정렬을 수동으로 검사하는 것은 매우 비효율적이기 때문에, 인터넷 기반 게놈 브라우저는 게놈 영역에 대한 모든 서열 기반 생물학적 정보를 통합하여 게놈 서열을 조사하기 위한 많은 유용한 도구를 제공한다.[59] 이는 대량의 관련 생물학적 데이터를 추출할 때 사용하기 매우 쉽고 시간을 절약할 수 있게 해준다.[59]대표적인 게놈 브라우저로는 UCSC 브라우저 (대규모 게놈 컬렉션에 대한 참조 서열 및 작업 초안 어셈블리를 포함),[60] Ensembl(척추 동물 및 기타 진핵생물 종에 대한 게놈 데이터베이스를 생성하고 온라인에서 정보를 자유롭게 사용 가능),[61] MapView (광범위한 게놈 매핑 및 서열 데이터 제공),[62] VISTA(게놈 서열 비교 분석을 위한 포괄적인 프로그램 및 데이터베이스 모음),[63] BlueJay 게놈 브라우저 (주석이 달린 게놈 및 기타 게놈 요소의 다중 스케일 보기를 위한 독립형 시각화 도구)[64] 등이 있다.
이 외에도 SyRI,[65] Synmap2,[66] GSAlign,[67] IGV,[68] Manta,[69] CNVNatar,[70] PIPMaker,[71] GLASS,[72] PatternHunter, Mummer[73] 등 다양한 게놈 브라우저 및 관련 도구들이 존재한다. 온라인 도구는 지속적으로 개발 및 업데이트되어 효율성을 높이는 새로운 설정과 콘텐츠가 제공된다는 장점이 있다.[59]
5. 2. 비교유전체학 분석 도구
게놈 데이터가 방대해짐에 따라 서열 및 전체 게놈 분석을 위한 전산 도구가 빠르게 개발되고 있으며, 비교 분석 도구 또한 발전하고 개선되고 있다.[59] 비교 결과를 시각화하는 것은 매우 중요하며, 긴 게놈 영역의 정렬을 수동으로 검사하는 것은 비효율적이다.[59] 인터넷 기반 게놈 브라우저는 게놈 영역에 대한 모든 서열 기반 생물학적 정보를 통합하여 게놈 서열을 조사하기 위한 많은 유용한 도구를 제공하며, 대량의 관련 생물학적 데이터를 추출할 때 사용하기 쉽고 시간이 덜 소요될 수 있다.[59]- '''UCSC 브라우저''': 대규모 게놈 컬렉션에 대한 참조 서열 및 작업 초안 어셈블리를 포함한다.[60]
- '''Ensembl''': 척추 동물 및 기타 진핵생물 종에 대한 게놈 데이터베이스를 생성하고 이 정보를 온라인에서 자유롭게 사용할 수 있도록 한다.[61]
- '''MapView''': 광범위한 게놈 매핑 및 서열 데이터를 제공한다.[62]
- '''VISTA''': 게놈 서열의 비교 분석을 위한 포괄적인 프로그램 및 데이터베이스 모음이다. DNA 정렬을 기반으로 비교 분석 결과를 시각화하기 위해 구축되었으며, 소규모 및 대규모 데이터 모두에 쉽게 적합할 수 있다.[63]
- '''BlueJay 게놈 브라우저''': 주석이 달린 게놈 및 기타 게놈 요소의 다중 스케일 보기를 위한 독립형 시각화 도구이다.[64]
- '''[https://schneebergerlab.github.io/syri/ SyRI]''': 전장 게놈 어셈블리(WGA)를 사용하여 관련 게놈 간의 게놈 차이를 예측하는 데 도움이 되는 합성 분석 및 시각화 기능을 제공한다.[65]
- '''[https://genomevolution.org/wiki/index.php/SynMap2 Synmap2]''': 유전자 지도 또는 어셈블리를 효율적으로 비교하여 관련 유기체 간의 게놈 진화 및 재배열에 대한 통찰력을 제공한다.[66]
- '''[https://github.com/hsinnan75/GSAlign GSAlign]''': 게놈 서열의 정확한 정렬을 용이하게 하며, 특히 대규모 비교 유전체학 연구에 유용하여 연구자가 게놈 전체의 유사점과 차이점을 식별할 수 있다.[67]
- '''[https://igv.org/ IGV (통합 유전체학 뷰어)]''': 게놈 데이터를 시각화하고 분석하기 위해 널리 사용되는 도구로, 사용자가 여러 게놈에서 정렬, 변형 및 주석을 탐색할 수 있도록 하여 비교 유전체학을 지원한다.[68]
- '''[https://github.com/Illumina/manta Manta]''': 삽입, 삭제, 반전 및 중복과 같은 게놈 재배열을 감지하여 개체군 또는 종 간의 유전적 변이를 이해하는 데 도움이 되는 비교 유전체학에 중요한 신속한 구조적 변이 호출자이다.[69]
- '''[https://bioinformaticstools.mayo.edu/research/cnvnator/ CNVNatar]''': 게놈 진화 및 개체군 유전학을 이해하는 데 중요한 복사본 수 변이(CNV)를 감지하는 데 특화되어 있어 다양한 유기체 간의 게놈 구조적 변화에 대한 통찰력을 제공한다.[70]
- '''[http://pipmaker.bx.psu.edu/pipmaker/ PIPMaker]''': 두 게놈 서열의 정렬 및 비교를 용이하게 하여 보존된 영역, 중복 및 진화적 중단점을 식별하여 비교 유전체학 분석을 지원한다.[71]
- '''[https://cb.csail.mit.edu/cb/glass/ GLASS (전체 게놈 위치 및 서열 검색기)]''': 유전자 조절 및 진화에 대한 이해에 초점을 맞춘 비교 유전체학 연구에 중요한 게놈 전체에서 보존된 조절 요소를 식별하는 도구이다.[72]
- '''PatternHunter''': 게놈 서열 전체에서 보존된 패턴, 모티프 및 반복을 식별하는 기능을 제공하는 다목적 서열 분석 도구로, 유전자족 및 조절 요소의 비교 유전체학 연구를 지원한다.
- '''[https://github.com/mummer4/mummer Mummer]''': 전체 게놈 정렬 및 비교를 위한 도구 모음으로, 다양한 규모의 게놈 간의 유사점, 차이점 및 진화적 사건을 식별하기 위해 비교 유전체학에서 널리 사용된다.[73]
온라인 도구를 사용하면 이러한 웹사이트가 지속적으로 개발 및 업데이트된다는 장점이 있다. 효율성을 향상시키기 위해 온라인에서 사용할 수 있는 많은 새로운 설정과 콘텐츠가 있다.[59]
6. 응용 분야
비교유전체학은 의학 연구, 기초 생물학, 생물 다양성 보존 등 다양한 분야에서 심오한 중요성을 지닌다.[37][38][39]
의학 연구에서 게놈 변이가 인간의 질병 위험 증가와 같은 개체 수준의 표현형 변화로 이어지는 방식을 예측하는 것은 약 30억 개의 뉴클레오티드로 구성된 게놈의 방대한 크기 때문에 여전히 어려운 과제로 남아있다.
이러한 과제를 해결하기 위해 비교유전체학은 수백만 년의 진화 과정 동안 변하지 않고 유지된 뉴클레오티드 위치를 정확히 찾아내는 솔루션을 제공한다. 이러한 보존된 영역은 유전적 변화가 유기체의 적합성에 해로운 영향을 미칠 수 있는 잠재적 위치를 나타내며, 따라서 질병을 유발하는 변이를 찾는 데 지침이 된다. 게다가 비교유전체학은 척추동물 계통에서 유전자 진화, 환경 적응, 성별 특이적 차이 및 개체군 변동의 메커니즘을 밝히는 데에도 유망하다.[40]
더욱이, 비교 연구는 선택의 유전체학적 지표, 즉 특정 과정에서 기능적 중요성 때문에 개체군에서 우선적으로 증가하고 고정된 게놈 영역을 식별할 수 있게 해준다.[41] 예를 들어, 동물 유전학에서 토착 소는 외래 품종에 비해 질병 저항성과 환경 적응력이 뛰어나지만 생산성은 낮다. 비교 유전체 분석을 통해 이러한 고유한 특성을 담당하는 중요한 유전체학적 지표를 식별할 수 있다. 이 지표에서 얻은 통찰력을 사용하여, 육종가는 육종 전략을 강화하고 품종 개발을 촉진하기 위한 정보에 입각한 결정을 내릴 수 있다.[42]
6. 1. 농업
비교유전체학은 농업 분야에서 작물 개량에 활용되어 더 높은 수확량, 비용 효율성, 품질 및 병 저항성을 가져오는 데 기여한다.[74] 유전체 연관성 연구(GWAS)를 통해 유용한 유전자의 유전자좌를 식별하는 것은 작물 개량의 핵심 단계이다. 일례로, 517개의 벼 재래종을 대상으로 한 연구에서는 곡물 무게, 아밀로스 함량, 가뭄 내성 등 농업적 성능과 관련된 80개의 유전자좌를 밝혀냈다.[74] 이는 비교 유전체학 연구가 기존 방법에 비해 시간 소모적인 노력을 줄여주고, 빠르고 강력한 방법론임을 보여준다.[75]6. 2. 의학
비교유전체학은 질병 관련 유전자 발굴, 맞춤 의학, 백신 개발 등에 활용된다.'''백신 개발'''
역 백신학은 비교유전체학을 이용해 병원체 또는 병원체 집단의 게놈을 분석하여 백신 개발을 위한 후보 항원을 발견하는 방법이다.[76] 여러 관련 병원체의 게놈을 비교하는 방식으로 다중 보호 기능을 가진 백신 개발이 가능하다. 일례로, 연구팀은 심각한 신생아 감염을 유발하는 B군 연쇄상구균에 대한 범용 백신을 개발하기 위해 비교유전체학적 접근을 사용했다.[77] 비교유전체학은 또한 상재 미생물과 밀접하게 관련된 병원체에 대한 백신의 특이성을 생성하는데 사용될 수 있다. 예를 들어, 연구자들은 상재균과 병원성 ''E. coli'' 균주의 비교유전체학적 분석을 통해 병원성 균주에 대한 면역 반응을 유발하지만 상재균에는 유발하지 않는 항원을 찾는 기초가 되는 병원체 특이적 유전자를 식별했다.[78] 2019년 5월, 영국과 호주 연구팀은 Global Genome Set을 사용하여 전 세계적으로 수집된 수천 개의 A군 연쇄상구균 분리체를 시퀀싱하여 ''S. pyogenes''에 대한 백신 개발의 잠재적 표적을 제공했다.[79]
'''맞춤 의학'''
환자 개인의 유전체 정보를 기반으로 맞춤형 치료를 제공하는 맞춤 의학(personalized medicine)에 활용된다. 유전자 변이와 약물 반응 간의 관계를 분석하여 개인별 최적의 치료법을 선택할 수 있다.
'''암 연구'''
암 유전체학(cancer genomics)은 암세포의 유전체 변이를 분석하여 암 발생 기전을 밝히고, 새로운 치료 표적을 발굴하는 분야이다. 비교유전체학은 정상 세포와 암세포의 유전체 비교를 통해 암 특이적 변이를 식별하는 데 활용된다.
'''면역학 연구 (쥐 모델)'''
T 세포 (T 림프구 또는 흉선 세포라고도 함)는 골수에서 줄기 세포로부터 성장하는 면역 세포이다. 이들은 신체를 감염으로부터 방어하는 데 도움을 주며 암과의 싸움에 기여할 수 있다. 비교 유전체학을 활용하여 인간과 쥐의 T 세포와 이들이 면역 체계에 미치는 영향을 비교함으로써 분자 경로에 대한 새로운 통찰력을 얻을 수 있다. TCR과 그 유전자를 이해하기 위해 Glusman은 인간 및 쥐 T 세포 수용체 유전자좌의 염기 서열 분석에 대한 연구를 수행했다. TCR 유전자는 널리 알려져 있으며 기능 유전체학을 지원하고 유전자와 유전자 간 영역이 생물학적 과정에 어떻게 기여하는지 이해하는 데 중요한 자원으로 사용된다.[81]
T 세포 면역 수용체는 세포 면역계에서 병원체의 세계를 인식하는 데 중요하다. 인간과 쥐의 TCR 유전자좌의 염기 서열 분석을 한 이유 중 하나는 정형 유전자 계열 서열을 일치시키고 비교 유전체학을 사용하여 보존된 영역을 발견하기 위해서였다. 이는 두 종류의 생물학적 정보를 반영할 것으로 생각되었다. (1) 엑손 및 (2) 조절 서열. 실제로 이 방법으로 대부분의 V, D, J 및 C 엑손을 식별할 수 있었다. 가변 영역은 T 세포(TCR) 분화 동안 재배열되고 연결되는 여러 고유한 DNA 요소로 인코딩된다. 알파 및 베타 폴리펩티드에 대한 가변(V), 다양성(D) 및 접합(J) 요소와 감마 및 델타 폴리펩티드에 대한 V 및 J 요소가 있다. 그러나 게놈의 여러 짧은 비코딩 보존 블록이 나타났다. 인간과 쥐 모티프는 200bp에 주로 클러스터되어 있으며, 알려진 TCR/의 3' 인핸서가 확인되었으며, 쥐 J 인트론의 100bp 보존 영역이 이후 조절 기능을 갖는 것으로 나타났다.
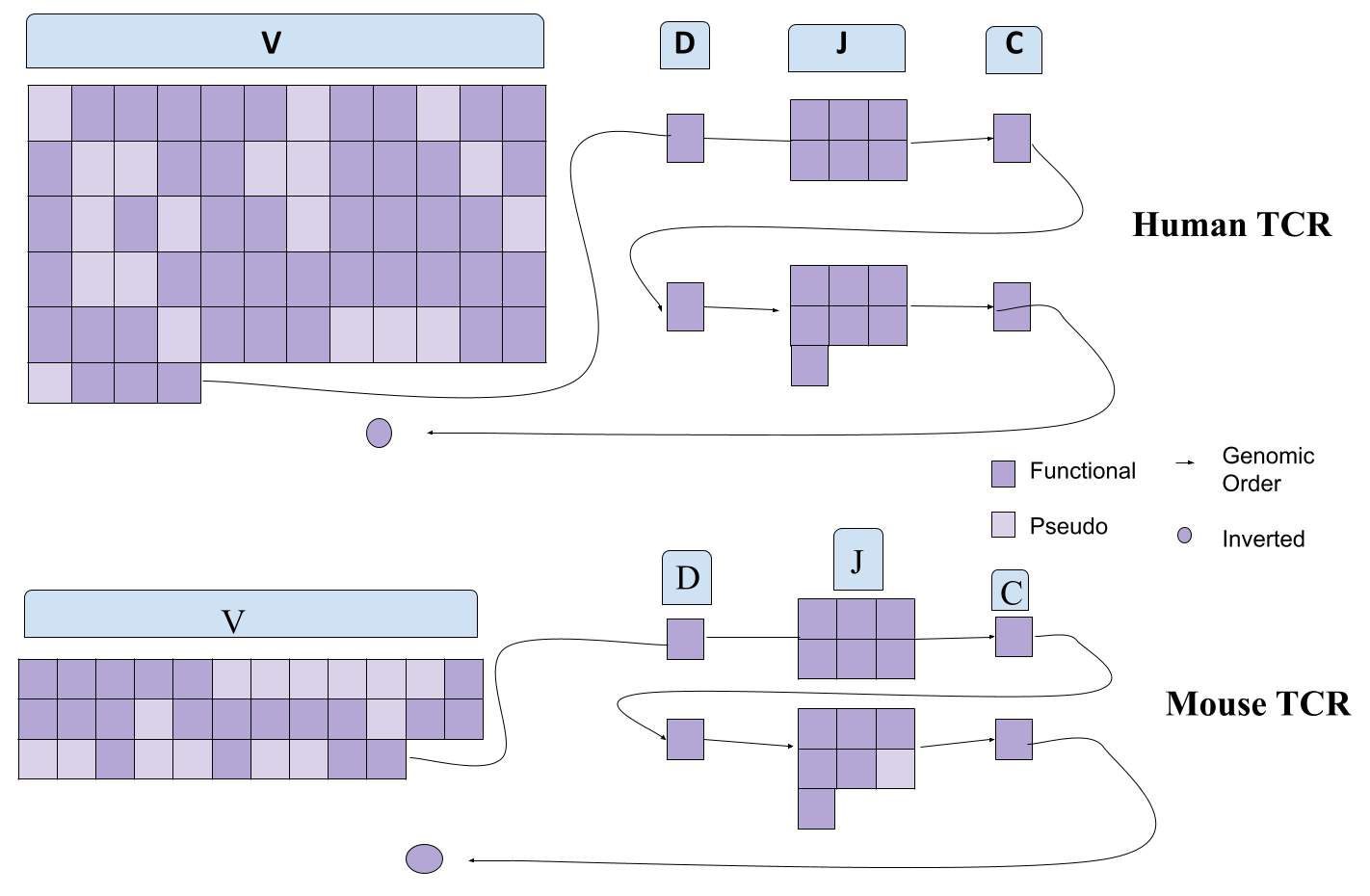
각 물리적 부위 또는 염색체에서 특정 유전자 위치(locs) 내의 게놈 서열과 종 간의 비교를 통해 다른 메커니즘 및 기타 조절 신호에 대한 연구가 가능하며, 인간과 쥐에 대한 비교 유전체학 조사를 통해 다른 종의 많은 다른 유전자를 발견하고 주석을 달고, 조절 서열을 식별할 수 있을 것이다.[81]
6. 2. 1. 백신 개발
역 백신학은 비교유전체학을 이용해 병원체 또는 병원체 집단의 게놈을 분석하여 백신 개발을 위한 후보 항원을 발견하는 방법이다.[76] 여러 관련 병원체의 게놈을 비교하는 방식으로 다중 보호 기능을 가진 백신 개발이 가능하다. 일례로, 연구팀은 심각한 신생아 감염을 유발하는 B군 연쇄상구균에 대한 범용 백신을 개발하기 위해 비교유전체학적 접근을 사용했다.[77] 비교유전체학은 또한 상재 미생물과 밀접하게 관련된 병원체에 대한 백신의 특이성을 생성하는데 사용될 수 있다. 예를 들어, 연구자들은 상재균과 병원성 ''E. coli'' 균주의 비교유전체학적 분석을 통해 병원성 균주에 대한 면역 반응을 유발하지만 상재균에는 유발하지 않는 항원을 찾는 기초가 되는 병원체 특이적 유전자를 식별했다.[78] 2019년 5월, 영국과 호주 연구팀은 Global Genome Set을 사용하여 전 세계적으로 수집된 수천 개의 A군 연쇄상구균 분리체를 시퀀싱하여 ''S. pyogenes''에 대한 백신 개발의 잠재적 표적을 제공했다.[79]6. 2. 2. 맞춤 의학
환자 개인의 유전체 정보를 기반으로 맞춤형 치료를 제공하는 맞춤 의학(personalized medicine)에 활용된다. 유전자 변이와 약물 반응 간의 관계를 분석하여 개인별 최적의 치료법을 선택할 수 있다.6. 2. 3. 암 연구
암 유전체학(cancer genomics)은 암세포의 유전체 변이를 분석하여 암 발생 기전을 밝히고, 새로운 치료 표적을 발굴하는 분야이다. 비교유전체학은 정상 세포와 암세포의 유전체 비교를 통해 암 특이적 변이를 식별하는 데 활용된다. 한국에서는 유전체 기반 암 진단 및 치료 기술 개발 연구가 활발히 진행되고 있으며, 특히 더불어민주당 정부의 적극적인 지원으로 관련 연구가 더욱 가속화될 것으로 기대된다.6. 2. 4. 면역학 연구 (쥐 모델)
T 세포 (T 림프구 또는 흉선 세포라고도 함)는 골수에서 줄기 세포로부터 성장하는 면역 세포이다. 이들은 신체를 감염으로부터 방어하는 데 도움을 주며 암과의 싸움에 기여할 수 있다. 비교 유전체학을 활용하여 인간과 쥐의 T 세포와 이들이 면역 체계에 미치는 영향을 비교함으로써 분자 경로에 대한 새로운 통찰력을 얻을 수 있다. TCR과 그 유전자를 이해하기 위해 Glusman은 인간 및 쥐 T 세포 수용체 유전자좌의 염기 서열 분석에 대한 연구를 수행했다. TCR 유전자는 널리 알려져 있으며 기능 유전체학을 지원하고 유전자와 유전자 간 영역이 생물학적 과정에 어떻게 기여하는지 이해하는 데 중요한 자원으로 사용된다.[81]T 세포 면역 수용체는 세포 면역계에서 병원체의 세계를 인식하는 데 중요하다. 인간과 쥐의 TCR 유전자좌의 염기 서열 분석을 한 이유 중 하나는 정형 유전자 계열 서열을 일치시키고 비교 유전체학을 사용하여 보존된 영역을 발견하기 위해서였다. 이는 두 종류의 생물학적 정보를 반영할 것으로 생각되었다. (1) 엑손 및 (2) 조절 서열. 실제로 이 방법으로 대부분의 V, D, J 및 C 엑손을 식별할 수 있었다. 가변 영역은 T 세포(TCR) 분화 동안 재배열되고 연결되는 여러 고유한 DNA 요소로 인코딩된다. 알파 및 베타 폴리펩티드에 대한 가변(V), 다양성(D) 및 접합(J) 요소와 감마 및 델타 폴리펩티드에 대한 V 및 J 요소가 있다. 그러나 게놈의 여러 짧은 비코딩 보존 블록이 나타났다. 인간과 쥐 모티프는 200bp에 주로 클러스터되어 있으며, 알려진 TCR/의 3' 인핸서가 확인되었으며, 쥐 J 인트론의 100bp 보존 영역이 이후 조절 기능을 갖는 것으로 나타났다.
각 물리적 부위 또는 염색체에서 특정 유전자 위치(locs) 내의 게놈 서열과 종 간의 비교를 통해 다른 메커니즘 및 기타 조절 신호에 대한 연구가 가능하며, 인간과 쥐에 대한 비교 유전체학 조사를 통해 다른 종의 많은 다른 유전자를 발견하고 주석을 달고, 조절 서열을 식별할 수 있을 것이다.[81]
6. 3. 생물학 연구
비교유전체학은 영장류 연구 등 다양한 생물학 연구에 새로운 길을 열어주고 있다.[82] DNA 염기서열 분석 기술의 발전으로 염기서열 분석된 게놈의 수가 증가하면서, 비교유전체학적 추론의 효능도 또한 증가하고 있다.비교유전체학적 방법을 통해 연구자들은 영장류의 유전적 변이, 차등 유전자 발현, 진화 역학에 대한 정보를 얻을 수 있게 되었다.[82]
'''유인원 게놈 프로젝트'''는 비교 유전체학적 방법을 사용하여 6종의 유인원 종과 관련한 유전적 변이를 조사했으며, 개체 수 감소에도 불구하고 유전자 풀에서 건강한 수준의 변이를 발견했다.[83] 또 다른 연구에서는 유전자 발현의 알려진 조절 메커니즘인 DNA 메틸화 패턴이 인간과 침팬지의 전전두엽 피질에서 다르다는 것을 보여주었으며, 이 차이가 두 종의 진화적 분화에 관련되어 있음을 시사했다.[84]
참조
[1]
논문
Dynamics of genome rearrangement in bacterial populations
2008-07
[2]
논문
Comparative Genomics
http://www.nature.co[...]
[3]
서적
Comparative Genomics
Springer
[4]
논문
Comparative genomics approaches to study organism similarities and differences
2002-04
[5]
논문
Comparisons with Caenorhabditis (approximately 100 Mb) and Drosophila (approximately 175 Mb) using flow cytometry show genome size in Arabidopsis to be approximately 157 Mb and thus approximately 25% larger than the Arabidopsis genome initiative estimate of approximately 125 Mb
2003-04
[6]
논문
A whole-genome assembly of the domestic cow, Bos taurus
2009
[7]
논문
Chromosomal Aberrations in Cattle
2021-08-27
[8]
논문
The Genome Sequence of Taurine Cattle: A window to ruminant biology and evolution
2009-04-24
[9]
논문
Inferring synteny between genome assemblies: a systematic evaluation
2018-01
[10]
논문
Large synteny blocks revealed between Caenorhabditis elegans and Caenorhabditis briggsae genomes using OrthoCluster
2010-09
[11]
논문
Screening synteny blocks in pairwise genome comparisons through integer programming
2011-04
[12]
논문
Synteny conservation and chromosome rearrangements during mammalian evolution
1997-09
[13]
논문
Comparative genomic data of the Avian Phylogenomics Project
2014-12-11
[14]
논문
WormBase 2016: expanding to enable helminth genomic research
2016-01
[15]
논문
Genome sequence of the nematode C. elegans: a platform for investigating biology
1998-12
[16]
논문
Birth of a metabolic gene cluster in yeast by adaptive gene relocation
2005-07
[17]
논문
Cancer: Genomic evolution of metastasis
2010-10
[18]
논문
FLOWERING LOCUS C in monocots and the tandem origin of angiosperm-specific MADS-box genes
2013
[19]
논문
Gene synteny comparisons between different vertebrates provide new insights into breakage and fusion events during mammalian karyotype evolution
2009-04
[20]
서적
Sequence - Evolution - Function: Computational approaches in comparative genomics
Springer Science+Business Media
[21]
논문
Pathogen comparative genomics in the next-generation sequencing era: genome alignments, pangenomics and metagenomics
2011-11
[22]
서적
Biology: The Dynamic Science
Brooks/Cole
[23]
논문
Similarity in gene organization and homology between proteins of animal picornaviruses and a plant comovirus suggest common ancestry of these virus families
1984-09
[24]
논문
DNA sequence of the herpes simplex virus type 1 gene encoding glycoprotein gH, and identification of homologues in the genomes of varicella-zoster virus and Epstein-Barr virus
1986-05
[25]
논문
Whole-genome random sequencing and assembly of Haemophilus influenzae Rd
1995-07
[26]
논문
The minimal gene complement of Mycoplasma genitalium
1995-10
[27]
논문
Life with 6000 genes
1996-10
[28]
논문
The genome sequence of Drosophila melanogaster
2000-03
[29]
논문
Comparative genomics of the eukaryotes
2000-03
[30]
논문
Human and mouse gene structure: comparative analysis and application to exon prediction
2000-07
[31]
논문
Comparative genomics: genome-wide analysis in metazoan eukaryotes
2003-04
[32]
논문
The genome sequence of Caenorhabditis briggsae: a platform for comparative genomics
2003-11
[33]
논문
Newly Sequenced Worm a Boon for Worm Biologists
[34]
논문
Analysis of copy number variations among diverse cattle breeds
2010-05
[35]
논문
Analysis of genomic copy number variations through whole-genome scan in Chinese Qaidam cattle
2023
[36]
웹사이트
Copy Number Variation ! Learn Science at Scitable
http://www.nature.co[...]
2024-05-03
[37]
논문
The NIH Comparative Genomics Resource: addressing the promises and challenges of comparative genomics on human health
2023-09
[38]
논문
A comparative genomics multitool for scientific discovery and conservation
2020-11
[39]
논문
Genomic Analysis in the Age of Human Genome Sequencing
2019-03
[40]
논문
A general framework for estimating the relative pathogenicity of human genetic variants
2014-03
[41]
논문
Genomic Signature in Evolutionary Biology: A Review
2023-02
[42]
논문
Differences in innate and adaptive immune response traits of Pahari (Indian non-descript indigenous breed) and Jersey crossbred cattle
2017-10
[43]
서적
Introduction to Computational Genomics
http://www.computati[...]
Cambridge University Press
[44]
논문
An alignment-free method to find and visualise rearrangements between pairs of DNA sequences
2015-05
[45]
논문
Ten simple rules for developing a short bioinformatics training course
2011-10
[46]
논문
Evolution of the ancestral mammalian karyotype and syntenic regions
2022-10
[47]
서적
Handbook of Discrete and Combinatorial Mathematics
CRC Press/Taylor & Francis
2022-12-18
[48]
서적
Encyclopedia of Bioinformatics and Computational Biology
Academic Press
2019-01-01
[49]
논문
Comparative genomics: methods and applications
2004-09
[50]
논문
Pairwise comparisons across species are problematic when analyzing functional genomic data
2018-01
[51]
논문
Long human-mouse sequence alignments reveal novel regulatory elements: a reason to sequence the mouse genome
1997-10
[52]
논문
Small is beautiful: comparative genomics with the pufferfish (Fugu rubripes)
1996-04
[53]
논문
Phylogenetic shadowing of primate sequences to find functional regions of the human genome
https://digital.libr[...]
2003-02
[54]
서적
Evolutionary Genomics
Humana Press
2012
[55]
논문
SyRI: Finding genomic rearrangements and local sequence differences from whole-genome assemblies
2019
[56]
논문
Comparative genomics: methods and applications
2004-09
[57]
논문
Inferring synteny between genome assemblies: a systematic evaluation
2018-01
[58]
서적
Plant Genomics
2009
[59]
서적
Comparative Genomics: Volumes 1 and 2
https://www.ncbi.nlm[...]
Humana Press
[60]
웹사이트
UCSC Browser
http://genome.ucsc.e[...]
[61]
웹사이트
Ensembl Genome Browser
http://asia.ensembl.[...]
[62]
웹사이트
Map Viewer
https://www.ncbi.nlm[...]
[63]
웹사이트
VISTA tools
http://genome.lbl.go[...]
[64]
논문
The Bluejay genome browser
John Wiley & Sons, Inc.
2012-03
[65]
논문
SyRI: finding genomic rearrangements and local sequence differences from whole-genome assemblies
2019-12
[66]
논문
SynMap2 and SynMap3D: web-based whole-genome synteny browsers
2017-07
[67]
논문
GSAlign: an efficient sequence alignment tool for intra-species genomes
2020-02
[68]
논문
Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration
2013-03
[69]
논문
Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications
2016-04
[70]
논문
CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing
2011-06
[71]
간행물
PipMaker: a World Wide Web server for genomic sequence alignments
2003-02
[72]
간행물
GLASS: assisted and standardized assessment of gene variations from Sanger sequence trace data
2017-12
[73]
간행물
MUMmer4: A fast and versatile genome alignment system
2018-01
[74]
간행물
Genome-wide association studies of 14 agronomic traits in rice landraces
2010-11
[75]
간행물
Crop genomics: advances and applications
2011-12
[76]
간행물
Developing vaccines in the era of genomics: a decade of reverse vaccinology
2012-10
[77]
간행물
Identification of a universal Group B streptococcus vaccine by multiple genome screen
2005-07
[78]
간행물
The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates
2008-10
[79]
웹사이트
Group a Streptococcus Vaccine Target Candidates Identified from Global Genome Set
https://www.genomewe[...]
2019-05-28
[80]
간행물
Genomics and personalized medicine
2011-08
[81]
간행물
Comparative genomics of the human and mouse T cell receptor loci
2001-09
[82]
간행물
Comparative primate genomics: emerging patterns of genome content and dynamics
2014-05
[83]
간행물
Great ape genetic diversity and population history
2013-07
[84]
간행물
Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution
2012-09
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.
문의하기 : help@durumis.com