가족성 샘종 폴립증
"오늘의AI위키"의 AI를 통해 더욱 풍부하고 폭넓은 지식 경험을 누리세요.
1. 개요
가족성 샘종 폴립증(FAP)은 APC 유전자 변이로 인해 발생하는 상염색체 우성 유전 질환으로, 수백에서 수천 개의 대장 폴립이 발생하여 대장암으로 발전할 수 있다. 소화기 증상 외에도 데스모이드 종양, 망막 색소 병변, 골종, 피지 낭종 등의 증상이 나타날 수 있으며, 가족력이 있거나 유전자 검사로 위험이 확인된 경우 대장 내시경, 유전자 검사 등을 통해 진단한다. 치료는 대장암 발생을 예방하기 위한 수술적 치료가 기본이며, 약물 치료와 정기적인 추적 관찰이 중요하다. 수술 후에도 폴립 재발 및 새로운 폴립 발생 위험이 있으므로 정기적인 추적 관찰이 필수적이다.
더 읽어볼만한 페이지
- 유전성 암 - 유전성 비폴립 대장암
유전성 비폴립 대장암은 DNA 불일치 복구 기작 결함으로 발생하는 유전 질환으로, 대장암을 포함한 다양한 암 발생 위험을 증가시키며, MLH1, MSH2, MSH6, PMS2, EPCAM 유전자 등의 돌연변이와 관련된다. - 유전성 암 - 남성유방암
남성유방암은 남성에게 드물게 발생하는 유방암으로, 여성 유방암과 차이를 보이며, 유방의 관에서 주로 발생하고 멍울, 유두 분비 등의 증상을 보일 수 있으며, 유방 촬영술, 초음파 검사, 조직 검사 등으로 진단하고 수술, 방사선 치료, 화학 요법 등으로 치료하며, 유전적 요인, 클라인펠터 증후군, 과도한 에스트로겐 수치, 음주 등이 위험 요인으로 작용한다. - 양성 종양 - 혈관종
혈관종은 혈관 내피세포 증식으로 생기는 양성 종양으로, 발생 시기, 형태, 위치에 따라 여러 유형으로 나뉘며, 임상적 진단 후 자연 퇴행을 기다리거나 약물 또는 외과적 치료를 시행한다. - 양성 종양 - 자궁근종
자궁근종은 가임기 여성에게 흔히 발생하는 자궁 평활근의 양성 종양으로, 위치와 크기에 따라 다양한 증상을 유발하거나 무증상일 수 있으며, 비정상 자궁 출혈, 월경 과다, 골반 통증 등이 주된 증상이고, 골반 검사, 초음파 검사, MRI 등을 통해 진단하며, 약물 치료, 수술, 자궁 동맥 색전술, 고강도 집속 초음파 치료 등 다양한 치료법이 활용된다. - 암 - 피부암
피부암은 피부 세포에서 발생하는 악성 종양으로, 자외선 노출, 유전적 요인, 면역 저하 등이 원인이 되며 기저세포암, 편평상피세포암, 흑색종으로 나뉘고 조기 발견과 자외선 차단이 중요하다. - 암 - 자궁경부암
자궁경부암은 인유두종바이러스 감염이 주원인인 자궁 경부의 악성 종양으로, 초기에는 증상이 미미하지만 진행 시 질 분비물 증가, 성교통, 골반통이 나타날 수 있으며, 자궁경부 세포진 검사, 조직 생검 등으로 진단하고 수술, 방사선 치료, 항암 치료 등으로 치료하며, HPV 백신 접종과 정기 검진으로 예방이 가능하다.
2. 원인
가족성 샘종 폴립증은 ''APC'' 유전자 변이로 인해 발생하며, 상염색체 우성 방식으로 유전된다. 변이된 유전자 하나만으로도 질환이 발생하며, 악성 종양 발생률은 100%에 육박한다. 환자는 대부분 이 질환을 가진 부모에게서 유전자를 물려받는다.[1]
2. 1. APC 유전자 변이
''APC'' 유전자의 변이는 상염색체 우성 방식으로 유전되며, 변이된 유전자 사본 하나만으로도 질환을 일으키기에 충분하다. 이러한 경우 악성 종양 발생률은 100%에 육박한다. 대부분의 경우, 영향을 받은 사람은 이 질환을 가진 부모 중 한 명을 둔다.[1]''APC''는 종양 억제 유전자로, 종양 발생을 예방하는 "문지기" 역할을 하는 다기능성 종양 억제 단백질인 샘종 폴립증(APC)을 생성하는 데 관여한다. APC 유전자의 결함은 APC가 제대로 기능을 하지 못함을 의미하며, 시간이 지남에 따라 APC에 의해 통제되어야 할 일부 세포가 통제되지 않고 계속해서 발달하여 암으로 변할 가능성이 있다. 가족성 폴립증의 경우, 이러한 현상은 일반적으로 소화관 표면에 폴립으로 나타난다.
폴립은 본질적으로 양성이지만, 투-히트 가설의 첫 번째 단계인 유전된 APC 돌연변이가 이미 발생한 상태이다. 종종 남아있는 "정상" 대립 유전자가 돌연변이를 일으키거나 삭제되어 폴립 생성이 가속화된다. APC 돌연변이 세포에서 추가적인 돌연변이(예: p53 또는 ''kRAS'')가 발생하면, 돌연변이가 없는 상피 세포보다 암으로 발전할 가능성이 훨씬 더 높다.
APC 유전자 산물의 정상적인 기능은 여전히 연구 중이며, 세포 핵과 막에 모두 존재한다. APC의 전형적인 종양 억제 기능은 β-카테닌 억제이지만, APC의 다른 종양 억제 기능은 세포 부착 및 세포 골격 조직과 관련될 수 있다.
''APC''의 돌연변이는 대장암 발생 사례에서도 흔히 발생하며, 이 암에서 APC의 중요성을 강조한다. 상염색체 우성 유전 유전 질환이며, 원인 유전자는 APC 유전자임이 밝혀졌다.
2. 2. MUTYH 유전자 변이
MUTYH는 DNA 수선 효소인 MYH 글리코실라제를 암호화한다. 정상적인 세포 활동 동안 구아닌은 때때로 산소에 의해 변형되어 시토신 대신 아데닌과 짝을 이루게 된다. MYH 글리코실라제는 이러한 실수를 염기 절제 복구를 통해 수정하여, 돌연변이가 DNA에 축적되어 종양 형성을 유발하지 않도록 한다. MYH 글리코실라제가 제대로 작동하지 않으면 DNA 오류가 축적되어 APC 돌연변이가 있는 환자와 유사한 임상 양상으로 종양 형성을 시작할 수 있다.MUTYH 유전자의 돌연변이는 상염색체 열성 패턴으로 유전된다. 이는 질환의 영향을 받으려면 유전자 사본 두 개가 모두 변경되어야 함을 의미한다. 대부분의 경우, 상염색체 열성 질환이 있는 어린이의 부모는 영향을 받지 않지만 변경된 유전자 사본을 하나씩 가지고 있는 보인자이다.
3. 유전 양상
가족성 샘종 폴립증은 다양한 유전 양상과 다양한 유전적 원인을 가질 수 있다. 이 질환이 APC 유전자의 변이로 인해 발생하는 경우, 상염색체 우성 방식으로 유전된다. 이는 변이된 유전자 사본 하나만으로도 질환을 일으키기에 충분하다는 것을 의미한다. 이러한 경우 악성 종양 발생률은 100%에 육박한다.[6] 대부분의 경우, 영향을 받은 사람은 이 질환을 가진 부모 중 한 명을 둔다.
NCBI에 따르면, APC 관련 용종증 진단을 받은 대부분의 개인에게는 영향을 받은 부모가 있지만, 가족력에서 질환이 인지되지 않거나, 증상 발현 전에 부모가 사망했거나, 영향을 받은 부모에게 질병이 늦게 발병하여 가족력이 음성으로 나타날 수 있다.[2] 또한, 약 20%의 사례는 ''드 노보'' 돌연변이이며, 겉보기상 드 노보 APC 돌연변이(즉, 알려진 가족력이 없음)가 있는 환자 중 20%는 체성 모자이시즘을 가지고 있다.[8] 무증상 개인(및 따라서 무증상 가족 구성원)도 존재하는 것으로 알려져 있다.[2]
상염색체 우성 유전 유전 질환이며, 원인 유전자는 APC 유전자임이 밝혀졌다.
4. 증상
가족성 샘종 폴립증(FAP)은 사춘기 초기부터 대장 폴립이 점차적으로 발생하는 질환이다. 초기에는 대부분 증상이 없지만, 시간이 지나면서 수백에서 수천 개의 폴립이 대장 표면에 발생한다.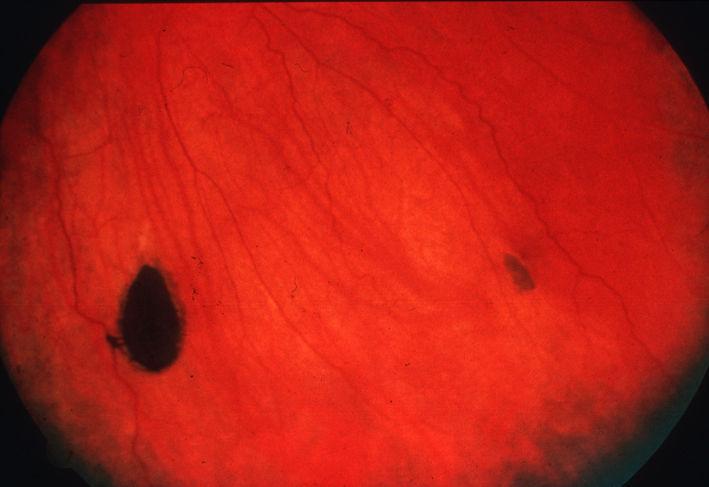
FAP는 십이지장 폴립을 동반할 수 있으며, 대장 내시경을 통해 진단한다.[1] FAP는 가드너 섬유종, 데스모이드 종양, 망막 색소 병변(CHRPE), 턱 낭종, 피지 낭종, 골종과 같은 소화기 외 증상을 동반할 수 있다.[4] 골종, 섬유종, 피지 낭종을 동반한 폴립증은 가드너 증후군으로 불린다.[5]
4. 1. 소화기 증상
가족성 샘종 폴립증 환자는 사춘기 초부터 수백에서 수천 개의 대장 폴립(때로는 다른 곳의 폴립)이 점차적으로 발생한다. 이러한 폴립은 출혈을 일으켜 변에 혈액이 섞여 나올 수 있다. 혈액이 보이지 않더라도 환자는 점진적으로 진행되는 철분 결핍으로 인해 빈혈이 발생할 수 있다.[1] 악성 종양이 발생하면 체중 감소, 배변 습관의 변화가 나타날 수 있다. 가족성 샘종 폴립증은 대장암으로 진행되기 전까지 거의 또는 전혀 징후를 보이지 않을 수 있다.수백 개에서 수만 개의 폴립이 발생한다. 폴립은 10세 전후에 발생하기 시작하여 시간이 지남에 따라 수와 크기가 증가한다. 이 폴립에서 대장암이 발생한다. 15세 전후부터 발생이 관찰되며, 40세에는 50%, 60세에는 거의 100%의 환자에게 대장암이 발생한다. 특이한 증상은 없으며, 혈변이나 설사가 발생하는 정도이다.
4. 2. 소화기 외 증상
골종이 턱뼈에 발생한다. 이는 소화관 폴립증 중에서도 가족성 샘종 폴립증(FAP)에 특이적인 것이다.[2]그 외에 갑상선암이나 췌장암의 합병도 보인다.[2]
5. 진단
가족성 샘종 폴립증(FAP)은 대장암 발생 전에 진단하는 것이 매우 중요하다. 이는 환자 본인뿐만 아니라, 질환의 영향을 받을 수 있는 다른 가족 구성원에게도 중요하다. FAP 진단에는 크게 대장 내시경 검사와 유전자 검사 두 가지 방법이 사용된다.
FAP 진단이 내려지면 대장 내시경을 통한 면밀한 추적 관찰과 폴립 절제술이 필요하다.
5. 1. 대장 내시경 검사
대장 내시경 검사는 돌연변이가 약화된 가족성 샘종 폴립증(FAP)의 경우 일반적인 진단 검사로 선택된다. 이는 폴립이 오른쪽 대장에 흔하게 위치하는데, S상 결장경 검사보다 대장 내시경 검사가 이를 더 잘 관찰할 수 있기 때문이다.[1] 대장 내시경 검사를 통해 다음과 같은 사항들을 확인할 수 있다.- '위험' 개인의 실제 임상 양상 및 상태 변화
- 대장 전체의 폴립 정량화 (폴립의 유무, 개수, 크기, 위치 등)
- 조직학적 진단 (세포/암 유형 감지)
- 폴립이 있는 경우 외래 환자 절제술(제거) 가능 여부 또는 수술 권장 여부
바륨 관장 및 가상 대장 내시경 검사 (의료 영상의 한 형태)도 FAP 진단을 제안하는 데 사용될 수 있다.
5. 2. 유전자 검사
유전자 검사는 95%의 경우에서 가족성 샘종 폴립증(FAP)의 최종 진단을 제공한다. FAP가 진단된 가족의 경우, 일반적으로 유전 상담이 필요하다.[1] 유전자 검사는 개인이 FAP에 취약한지, 혹은 결함 있는 APC 유전자를 상속했는지 여부를 보여줄 수 있다. 그러나 환자의 실제 상태는 유전자 검사로 결정할 수 없으며, 이는 직접적인 신체 검사를 통해서만 확인할 수 있다.미국 국립생물공학정보센터(NCBI)는 1997년에 "FAP에 대해 평가받은 개인의 거의 3분의 1에서 의사가 검사 결과를 잘못 해석했다"고 언급하며, 모든 유전자 검사의 "위험, 이점 및 제한 사항"을 의사가 이해해야 한다고 명시한다.[7]
질병을 유발하는 돌연변이가 확인된 가족 구성원의 경우, 산전 검사가 가능하다. 하지만 일반적으로 성인기에 발병하는 질환에 대한 산전 검사는 드물며, 신중한 유전 상담이 필요하다.
가족 중에 FAP 환자가 있는 경우에는 조기에 검사를 실시하여 최대한 빠른 진단과 치료가 이루어지도록 한다.
5. 3. 영상 검사
복부 의료 초음파 및 혈액 검사를 통해 간 기능 검사를 평가하여 간으로의 전이 여부를 확인한다.[7] 턱뼈에 골종이 발생하는 경우가 있는데, 이는 소화관 폴립증 중에서도 가족성 샘종 폴립증에 특이적인 소견이다.[7] X선 검사를 통해 다른 증상이 있는지 추가적으로 확인하며, 갑상선암이나 췌장암 등의 합병증도 나타날 수 있다.6. 치료
가족성 샘종 폴립증의 치료 목표는 대장암 발생을 예방하고, 합병증을 관리하는 것이다. 임상 관리는 FAP 위험이 있는 개인을 식별하고, FAP 유무를 진단하며, 선별/모니터링 프로그램을 통해 장관의 건강 상태를 확인하는 것을 포함한다.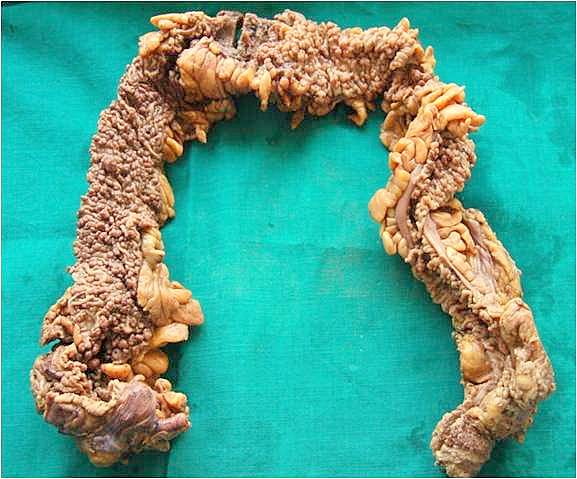
폴립의 악성 변성을 늦추기 위해 비스테로이드성 소염제(NSAIDs)를 비롯한 다양한 약물이 연구되고 있다. NSAIDs는 폴립의 수를 현저히 감소시키는 것으로 나타났지만, 추적 관찰하고 내시경적으로 치료하기에는 폴립이 너무 많기 때문에 일반적으로 관리에 큰 변화를 주지는 않는다. 오르니틴 탈탄산 효소의 억제제인 에플로르니틴은 트리파노소마증 치료에 사용되며, FAP 치료를 위해 NSAID 세레콕시브와 함께 잠재적인 예방 약물로 연구되고 있다. 또 다른 연구 약물은 NSAID와 함께 사용되는 설린닥이다.[9][10][11]
6. 1. 수술적 치료
가족성 샘종 폴립증(FAP)의 치료법은 유전자형에 따라 다르다. APC 돌연변이 환자의 대부분은 40세가 되기 전에 결장암이 발생하지만, 덜 흔한 약화된 형태는 일반적으로 늦은 나이(40~70세)에 나타난다. 따라서, 많은 경우 25세 이전에, 또는 적극적인 모니터링으로 발견 시 예방적 수술이 권장될 수 있다. 결장 또는 결장과 직장을 모두 제거하는 몇 가지 수술 방법이 있다. 100개 이상의 폴립이 있거나, 심각한 이형성 폴립이 있거나, 1cm보다 큰 폴립이 여러 개 있는 경우, 예방적 결장절제술이 필요하다.[9][10][11]수백 개에서 수만 개의 폴립이 발생하며, 이 폴립에서 대장암이 발생한다. 40세에는 50%, 60세에는 거의 100%의 환자에게 대장암이 발생한다.
대장암 발생을 예방하기 위해 기본적으로 대장 점막을 전부 절제한다. 일반적으로는 전체 결장과 직장 점막을 절제하고, 회장 낭 항문 문합술을 시행하지만, 하부 직장이나 항문관에 암이 존재하는 경우에는 직장도 전부 절제한다. 수술은 암 발생이 드문 20세 이전에 시행한다.
- 직장 침범: 직장과 결장의 일부 또는 전부를 제거한다. 환자는 회장루 (변이 복부의 가방으로 들어가는 영구적인 인공 항문)가 필요하거나 회장-항문 주머니 재건술을 받을 수 있다. 직장 제거 결정은 직장의 폴립 수와 가족력을 고려하여 결정한다. 직장에 폴립이 거의 없는 경우, 결장의 일부 또는 전부를 제거하고 소장(회장)을 직장에 직접 연결할 수 있다 (회장직장 문합술).
- 직장 비침범: 폴립이 나타나는 결장 부위를 제거하고 양쪽 끝을 다시 연결할 수 있다 (부분 결장절제술). 이 수술은 상당한 회복 기간이 필요하지만 삶의 질은 대체로 유지된다.
6. 2. 약물 치료
비스테로이드성 소염제(NSAIDs)는 폴립의 수를 줄이는 것으로 알려져 있다.[9][10][11] 오르니틴 탈탄산 효소 억제제인 에플로르니틴은 트리파노소마증 치료에 사용되지만, FAP 치료를 위해 NSAID인 세레콕시브나 설린닥과 함께 사용되어 예방 약물로 연구되고 있다. 그러나 이러한 약물 치료는 수술을 대체할 수는 없으며, 보조적인 치료 방법으로 활용된다.6. 3. 추적 관찰
가족성 샘종 폴립증(FAP) 환자는 정기적인 추적 관찰이 필요하다. 여기에는 다음이 포함된다.- 정기적인 내시경 검사: 수술 후에도 남아있는 직장, 회장낭, 십이지장 등에 폴립이 재발하거나 새로 발생할 수 있으므로 정기적인 내시경 검사가 필요하다.[1]
- 상부 위장관 내시경 검사 (EGD): 위, 십이지장에 발생하는 선종 및 암 발생 여부를 확인하기 위해 시행한다.[1] 환자의 약 60%에서 위에 폴립이나 선종이 발생하며, 주로 위저선과 유문선에서 발생하지만 악성화되는 경우는 드물다. 십이지장에는 높은 빈도로 선종이 발생하며, 암으로 발전하는 경우도 있다.
- 외래 대장내시경: 1~3년에 한 번씩 시행하거나, 유전자 혈액 검사를 통해 감수성을 확인하거나 부정한다. 발견된 소수의 용종은 시술 중에 절제할 수 있지만, 더 심각한 징후나 많은 수의 용종이 있는 경우 입원 수술이 필요할 수 있다.
- 기타 검사: 주장 조영술, 대장 내시경 검사, 생검, X선 검사 등을 통해 다른 증상이 없는지 검사한다.
FAP는 유전 질환이므로, 가족 중에 FAP 환자가 있는 경우 조기에 검사를 실시하여 최대한 조기 진단 및 치료가 이루어지도록 해야 한다.
7. 합병증
가족성 샘종 폴립증(FAP) 환자는 사춘기 초기부터 수백에서 수천 개의 대장 폴립이 점차적으로 발생하며, 이는 대장암으로 진행될 수 있다. 대장 폴립은 출혈을 일으켜 혈변의 원인이 되거나, 철분 결핍으로 인한 빈혈을 유발할 수 있다. 악성 종양이 발생하면 체중 감소, 배변 습관 변화, 전이 등의 증상이 나타날 수 있으며, 암으로 진행되기 전까지는 징후가 거의 없을 수도 있다.[1]
FAP는 시간이 지나면서 매우 점진적으로 발달하므로, 폴립은 사춘기부터 노년기까지 어느 시점에서든 암으로 발전할 수 있다. 대장 내시경 검사를 통해 폴립을 조기에 발견하는 것이 중요하다.[1]
FAP의 유전적 요인은 십이지장, 위, 갑상선암, 췌장암 등 다른 악성 종양 발생 위험을 높인다.[4] 가드너 섬유종, 데스모이드 종양(양성 피부 종양), 망막의 색소 병변("CHRPE—선천성 망막 색소 상피 비대"), 턱 낭종, 피지 낭종, 골종 (양성 골 종양) 등의 징후가 나타날 수 있다. 폴립증, 골종, 섬유종, 피지 낭종의 조합은 가드너 증후군이라고 한다.[5]
대장암이 진행되기 전, 폴립이 장 내벽에 국한되어 있을 때 수술을 통해 암을 예방하거나 제거할 수 있다. 수술 후 부분 결장 절제술을 시행한 경우에도 남은 결장에 대한 대장 내시경 감시가 필요하다.
섬유종은 침윤적 성질과 생명 유지 구조에 근접할 가능성 때문에 FAP 환자에서 사망의 두 번째로 높은 원인이다.[12] 턱뼈에 골종이 발생하기도 하며, 이는 소화관 폴립증 중에서도 FAP에 특이적인 것이다.
8. 예후
가족성 샘종 폴립증(FAP)이 암 전 단계에서 발견 및 관리되거나 암성 용종이 여전히 장 내부에 있을 때, 수술은 재발 없이 암을 예방하거나 제거하는 데 매우 높은 성공률을 보인다. 이는 암을 유발하는 위치가 수술로 완전히 제거되기 때문이다.
부분 결장 절제술을 시행한 경우, 환자는 여전히 대장암 발생 위험이 있으므로 남은 결장에 대한 대장 내시경 감시가 필요하다. 그러나 이러한 일이 발생하면, 이는 원래 수술로 제거된 암의 재발이나 전이가 아닌, 수술 후 제거되지 않은 결장 부위에서 새롭게 발생하는 용종으로 인한 새로운 사건이 된다.
섬유종은 침윤적 성질과 생명 유지 구조에 대한 잠재적 근접성으로 인해 사망의 두 번째로 높은 원인이다.[12]
대장암 발생을 예방하기 위해 기본적으로 대장 점막을 전부 절제한다. 일반적으로는 전체 결장과 직장 점막을 절제하고, 회장 낭 항문 문합술을 시행하지만, 하부 직장이나 항문관에 암이 존재하는 경우에는 직장도 전부 절제한다. 수술은 암 발생이 드문 20세 이전에 시행한다.
9. 관련 질환
- 가드너 증후군: 가족성 샘종 폴립증(FAP)에 두개골이나 장관골의 골종 및 연부 조직 종양이 함께 나타나는 질환이다. 원인이 되는 유전자는 FAP와 마찬가지로 APC 유전자이다. 치료법은 FAP와 같다.
- 터코트 증후군: 폴립증에 중추 신경계 종양이 함께 나타나는 질환이다. FAP에 비해 대장의 선종은 적지만, 악성으로 변하는 것은 더 이른 나이에 나타나기 쉽다. 상염색체 열성 유전이며, 원인이 되는 유전자는 아직 밝혀지지 않았다. 대장 전절제술을 시행하며, 예후는 뇌종양에 따라 달라진다.
- 포이츠-예거스 증후군
참조
[1]
논문
Genotype-phenotype correlations in attenuated adenomatous polyposis coli
1998-06
[2]
간행물
GeneReviews NBK1345
[3]
웹사이트
Familial adenomatous polyposis: MedlinePlus Genetics
https://medlineplus.[...]
2023-06-09
[4]
논문
Soft Tissue Special Issue: Fibroblastic and Myofibroblastic Neoplasms of the Head and Neck
null
2020-03
[5]
논문
A genetic and clinical study of intestinal polyposis, a predisposing factor for carcinoma of the colon and rectum
1951-06
[6]
논문
A target-selected Apc-mutant rat kindred enhances the modeling of familial human colon cancer
[7]
논문
Phenotypic variability of familial adenomatous polyposis in 11 unrelated families with identical APC gene mutation
1994-06
[8]
논문
Somatic APC mosaicism: an underestimated cause of polyposis coli
http://gut.bmj.com/c[...]
2008-01
[9]
논문
An international randomised trial of celecoxib versus celecoxib plus difluoromethylornithine in patients with familial adenomatous polyposis
https://gut.bmj.com/[...]
2016-02
[10]
논문
Eflornithine plus Sulindac for Prevention of Progression in Familial Adenomatous Polyposis
2020-09-10
[11]
논문
Efficacy and safety of eflornithine (CPP-1X)/sulindac combination therapy versus each as monotherapy in patients with familial adenomatous polyposis (FAP): design and rationale of a randomized, double-blind, Phase III trial
2016-12
[12]
웹사이트
Desmoid Tumor - Risk Factors
https://www.cancer.n[...]
2023-08-19
[13]
웹사이트
Familial Adenomatous Polyposis
https://www.lecturio[...]
2021-07-22
[14]
논문
American Founder Mutation for Attenuated Familial Adenomatous Polyposis
2008-01
[15]
논문
"[Impact of a registry on the survival familial adenomatous polyposis.]"
[16]
웹사이트
대한민국 질병관리본부 - 가족성 용종증
http://www.cdc.go.kr[...]
본 사이트는 AI가 위키백과와 뉴스 기사,정부 간행물,학술 논문등을 바탕으로 정보를 가공하여 제공하는 백과사전형 서비스입니다.
모든 문서는 AI에 의해 자동 생성되며, CC BY-SA 4.0 라이선스에 따라 이용할 수 있습니다.
하지만, 위키백과나 뉴스 기사 자체에 오류, 부정확한 정보, 또는 가짜 뉴스가 포함될 수 있으며, AI는 이러한 내용을 완벽하게 걸러내지 못할 수 있습니다.
따라서 제공되는 정보에 일부 오류나 편향이 있을 수 있으므로, 중요한 정보는 반드시 다른 출처를 통해 교차 검증하시기 바랍니다.
문의하기 : help@durumis.com